Diagnosing EDS and HSD

This whole post (born in 2014) was nearly obsoleted entirely in March 2017 by the BRAND new ginormous EDS nosology (classification) and diagnostic criteria, which replaced BOTH the old Villefranche nosology AND the Brighton (with an “r”, not “e”) Diagnostic criteria.
But NOT the Beighton (with an “e”!) 9 pt hypermobility scale (or score) which was just part of the Brighton diagnostic criteria in a plot to confuse -that is still in use -for now. (Phew! Confusing!)
Update April 24, 2026: The newly defined Hypermobility Spectrum Disorders invented and carved out in 2017 by The Ehlers-Danlos Society are going to be recombined with hypermobile type EDS (hEDS) per the new diagnostic criteria and nosology coming out on December 2nd, 2026 now. The final name and categorization are still TBD.
Thanks for your patience while I continue updating my site to reflect all the changes. And please refer to this site via the international Ehlers-Danlos Society for the ultimate answers as they are dictating the rules. And, please know, this page reflects the last formally accepted nosology and diagnostic criteria put out in 2017, until they are supplanted in December 2026.
Note for doctors May 2018: The EDS Toolkit for General Practitioners originally produced and hosted by the Royal College of General Practitioners in May 2018, is now hosted by EDS UK since January 2022. Any doctor that is willing can technically diagnose the new HSDs and all forms EDS, but especially the most common form: hypermobile EDS (hEDS). It’s just that few are willing or have time do so as yet. But please be sure to do proper full differential diagnosis! (See below for more on that.)
You have my sympathy with the process. I fear the Toolkit above makes it all seem a little too simple, when in fact, it’s not if you do full proper differential diagnosis. (Rule out all the more rare forms of EDS, plus all the other similar connective tissue disorders like Osteogenesis Imperfecta, Loeys-Dietz, etc. See below.)
Some may still wish to refer more distinct or unusual cases to the geneticist right away and probably should. But the new nosology and the toolkit above were designed specifically to ease the burden on medical genetics offices. (Personal note: I was honored to be tapped by the project lead as a contributing author based on this blog and my Twitter stream.) Read on for a more in-depth description.
ETA 10/30/24: I just learned of a retired geneticist, Dr. Golder Wilson in the state of Texas (USA) who feels you do not need genetic testing to diagnose any form of EDS. That testing is not that accurate: you can have a SNP but not show signs of the matching disease. And vice-versa: show signs of a form of EDS, but not have the matching SNP for that form. This matches what I’ve heard from other geneticists in the past. But, you can go for testing if you really want, and especially if you do strongly suspect vascular EDS (vEDS). (See below for more on that.) You can also get help from him to be diagnosed remotely now. (He’s retired.) Keep this in mind as you plow through the following.
Last updated April 24, 2026
13 Types of EDS Newly Defined in 2017

Since it’s often so hard to get a doctor or doctors to listen to us and recognize our vast collections of symptoms as a possible systemic condition, we naturally want an easy test to take and be done with this painful journey. Our often ADHD brains and over-worked doctors want this too, natch.
Well, I’m sorry to break it to you, but there is no single test for ALL of the types of Ehlers-Danlos Syndromes yet.
And as of March 2017 the old diagnoses of HMS, JHS and BJHS have all been supplanted by both the new hEDS and HSD classification per The Ehlers-Danlos Society (TEDS). Not all doctors are up to date nor on board with this for various reasons. Direct questions to TEDS at info@ehlers-danlos.com. See also Road to 2026.
Update 10/1/24: We’ve just become aware that there is a single gene panel now available to run when a doctor suspects one or more of the rare genetic forms of EDS, (currently 12 of the 13 types recognized in 2024) versus needing to pick just one or two and run them separately. Check out the 34 minute mark of this podcast interview with Dr. Alan Hakim in 2024 for more on that. NB It is not meant to just be run for any- and everyone, but when you do suspect one or more truly rare forms EDS with known genetic markers. (ETA 10/30/24: Though Dr. Wilson in Texas still doesn’t think that’s truly necessary, but you can if you really want to.)
And there is no single tissue marker or genes identified yet to test for the most common type of EDS, known as hypermobile EDS or hEDS (formerly known as Type III EDS). Which The Society still insists on calling rare since they invented the Hypermobility Spectrum Disorders for all those who fail the hEDS checklist, though several of us beg to differ. However…
Additional update 10/1/24: We bring exciting news of a POTENTIAL biomarker just recently published in September 2024 that may be able to help diagnose hypermobile EDS (hEDS) and HSD in the future! This study still needs to be replicated and a commercial test developed, but the finding is quite distinct, and of course quite exciting after so many years of gaslighting and confusion for hEDS and HSD patients. (Stay tuned.)
Yes, whole genome (WGS) and whole exome sequencing (WES) tests are coming way down in price and thus in reach for more patients at just $500 or less. But, this is not a practical move. You end up with seriously overwhelming heaps of unnecessary data to weed and sort through, and the bench research to match it with is still not there.
I.e, you may have many interesting Variants of Unspecified Significance on genes of interest, aka “VUSes”, but no one may know what to do with them yet. Science needs a chance to catch up. Same goes for WES testing still too, though it is just a subset of your genome too. Please talk to a geneticist if you insist on doing this. (Dr. Hakim addresses this also in this podcast after the 34 minute mark.)
History of EDS Nosology and Diagnosis
There were six main types of EDS recognized until March 14th, 2017, but several one-off mutations in single families around the world with new ones found periodically. There are now 13 total types formally recognized as of March 2017, up from the former six main types prior. In May 2018 a really rare autosomal recessive gene was announced but not yet named or folded into the nosology. And possibly one other I’m missing here. (This will keep happening over time.)
And at least two more candidate genes to explain some cases of hEDS have been uncovered since 2023 (MIA3 and Kallikrein), though testing is not available for these yet, and they are also not yet part of the 2017 nosology. I’m sure more genes are waiting in the wings to be announced soon too, thanks all of you dedicated scientists and researchers. (See Road to 2026.)
Five of those six former types were considered rare, but now all thirteen formally recognized types of EDS (as of 2017) are considered rare by TEDS again based on the new criteria, and twelve of the thirteen now have known genetic markers. Only the most common type, hypermobile EDS, aka “hEDS” does not – yet. (Some candidate genes are arising slowly, and a fibronectin fragment was found in 2024 but is not yet available as a commercial test.)
SPOILER ALERT: Hypermobility Spectrum Disorders are a diagnosis of exclusion of ALL of the above, plus ALL other similar connective tissue diseases like Marfans, Loeys-Dietz, Sticklers, etc. So there is no genetic test for it. It’s a clinical diagnosis, still, period. (As of 10/1/24.) But ONLY AFTER PROPER FULL DIFFERENTIAL DIAGNOSIS is done! (And heads up, this will change again in December 2026 when the HSDs are folded back in with hEDS as announced in April 2026.)
In other words, before Dece 2, 2026, you have to diligently and thoroughly rule out ALL OTHER POSSIBLE FORMS OF EDS OR OTHER CTDs based on history and presentation. THEN you may diagnose a form of HSD. Not the other way around. Yes, that’s upwards of 20+ conditions to wade through. Again, my sympathies.
Suggested Diagnostic Flow Chart
| 1a – Do they seem like they have some kind of CTD? | 1b – Gather as much medical history as you can to see (I recommend using a spreadsheet) |
| 2a – Rule out all rare forms of EDS with genetic markers (12 of 13 as of 2024) if suspected | 2b – There are 12 types with markers per the 2017 nosology here |
| 3a – If negative by testing for those, then rule out all other possible similar CTDs like: | 3b – Marfans, Loeys-Dietz, Stickler Syndrome, Osteogenesis Imperfecta, Cutis Laxa |
| 4a – If negative for those, then rule out hEDS | 4b – No genetic test yet*, see hEDS Diagnostic Checklist |
| 5a – If negative for hEDS, then diagnose a form of Hypermobility Spectrum Disorder | 5b – This is a diagnosis of exclusion of ALL THE above! |
9/23/25: I was just alerted to this new hEDS and HSD self-screening tool for patients in AUSTRALIA ONLY this past weekend. Available to anyone in Australia with internet access, both patients and doctors can use it to help decide when to suspect either an HSD, hEDS or other form of EDS. (They would like to expand it outside of Australia someday but had to start there for now. Patience everyone!)
Diagnostic Issues
As noted, the most common type by far still, hypermobile EDS or hEDS as it’s called now (formerly “Type III”) does not have a genetic or biomarker yet, so it can only be diagnosed clinically still using the new 2017 diagnostic criteria for it.
As of March 2017: The criteria for hEDS were tightened a good bit, and the entirely new category of Hypermobility Spectrum Disorders or “HSD” was proposed and adopted to catch the rest of us who show some hypermobility, but no longer meet these new criteria. (You may be very bendy, but not have enough issues with your tissues to pass, or have lots of systemic trouble, but not be flexible or bendy enough to pass the hEDS criteria.)
Again heads up: this will be changing again on December 2nd, 2026 when they will fold the HSDs back in with hEDS. Unsure what it will be called, nor how catagorized ultimately. That remains to be seen.

And no, you cannot diagnose yourself via your raw data from 23andMe or Ancestry etc., contrary to rumors in many online support groups.
These are likely finding nonsense mutations or non-pathogenic single point mutations aka “SNPs” that aren’t known disease-causing mutations. Further, there can be other kinds of pathological mutations (duplications, deletions) that you can’t uncover this way.
Not all SNPs cause disease or trouble just because they are on a gene of interest (e.g. ADAMST2, PLOD1 etc.), some are just variations from the wild type, period, like hair color. You wouldn’t say brown-haired people have a hair disease for instance, right? The bench research needs to catch up. Please work with a medical doctor (ideally a geneticist) for proper diagnosis if you truly suspect a rare form of EDS, or similar connective tissue disease. (Marfans, Stickler’s, Osteogenesis Imperfecta, Cutis-Laxa, Loeys-Dietz.)
Few doctors will take such a self-diagnosis seriously, nor should they. One geneticist I know has found errors in his patient’s 23andMe data after running WES on them later. And the second-party data interpreters (LiveWello, Genetic Genie, etc.) are not known for accuracy for another thing.
I’ve gotten conflicting results from my own raw data after downloading and parsing it through the same third-party parser at different times. One time it said I had a particular defect, the other time it said I didn’t. From the same identical unaltered zip file. Which one is correct? I may never know. But I don’t think I should medicate or diagnose based on such questionable info! To quote Dr. Ben Lynch, “treat the person, not the SNP“, always.
Please do consult a qualified doctor if able to get the most accurate and complete diagnosis while ruling out other similar Heritable Connective Tissue Diseases (HCTDs) and other similar or secondary conditions while we continue tracking down more causes of hypermobile EDS. You might look for a local support group to help find the knowledgeable doctors in your region.
Hypermobility, often mis-interpreted to mean flexibility by most people itself is just a trait, like red hair color, and in itself is generally benign. It is thought to run in about 20 – 40% of the general population and lends nicely to gymnastic and dancing and singing careers.
The EDS and now also the HSDs all involve hypermobility plus symptoms, or issues like fallen arches, bad teeth, tendinitis, hernias, easy bruising, varicose veins and so much more that ultimately drive you to see the doctor a lot for those who aren’t in denial.
The New Diagnostic Pathway since 2017 (until December 2026)
First, Suspect a Connective Tissue Disorder
I highly recommend my When to Suspect and When Else To Suspect EDS and HSD pages for starters. Obviously, it helps if you can gather biological family history, but this is not always possible. (Some of us are adopted, others disowned or abandoned or our family is not cooperative.)
Fill out a spreadsheet if you can
Here is a sample spreadsheet for helping to gather some of your relevant family history in a fixed, unmodifiable sample PDF Here. This is just a starting example I developed, you can create your own using a spreadsheet program like Excel or Google documents as well. (October 2018 editor’s note: There used to be another great pre-diagnostic medical workup page linked from The Coalition Against Pediatric Pain here too, but it seems to have been deleted, sorry!)
Your geneticist will love you for this if you get to see one, as it really helps them to see patterns in your family history. I would fill one out one for each side of your family, as able. (E.g. one for mom’s, one for dad’s side, if known. List yourself and your siblings both times.)
If you do suspect a rare type, this may also help you get a referral to a geneticist if needed for proper diagnosis. And if not, it will help any willing doctor to diagnose you together. Don’t despair if you lack family history though. While quite helpful, lack of family medical history and living family members to examine should not stop a good diagnostician from succeeding.
And it’s okay if you can’t record or capture everything from everyone. (Don’t get hung up on achieving perfection.) Just record and note what you can as best you can. Even if you think it’s unrelated. You might be surprised.
Do proper differential diagnosis
Unfortunately, you are stuck wading through the descriptions of all of the 12 rare types of EDS with known genetic markers one by one to become acquainted with their hallmark traits, to know if you should suspect one. As well as signs of the similar connective tissue disorders to also rule out like Marfans, Loeys-Dietz Syndrome, Stickler Syndrome, Osteogenesis Imperfecta, and more. (So yeah, upwards of 20 or more conditions, sorry.) Some of these traits include things like:
- club foot/feet
- bilateral hip dysplasia (aEDS)
- extremely stretchy/saggy or wrinkly skin even when young (dEDS)
- distinct hypertrophic or atrophic scarring, or keloid scars (several types)
- extremely thin skin that tears easily (vEDS and cEDS and clEDS)
- blue sclerae in the eyes (vEDS and or cEDS)
- extreme early scoliosis or kyphosis (all types can have mild scoliosis) (kEDS)
- keratoconus (droopy corneas lending to astigmatism) – (sevearl types)
- uterine or other hollow organ ruptures or family history of same (vEDS)
- family history of early death from aneurysms, easy bleeding or organ ruptures (vEDS)
- Severe, early-onset periodontitis and or detached gingiva (pEDS)
- Short stature with muscle hypotonia and or contractures (spEDS)
- Aortic root dilation (especially in children – get a baseline) (several)
- Widely spaced eyes, bifid uvula, cleft palate, malar hypoplasia, and blue sclerae (Loeys-Dietz)
- Pectus excavatum or carnatum (sunken or pigeon chest) – (Marfans and Loeys-Dietz)
- Long slender fingers and or toes, marfanoid habitus (arm span to body length ratio) – see Marfans
- Lens dislocations – see Marfans
- Tortuous arteries and or veins and bleeding issues plus hypermobility – see Loeys-Dietz
- Extreme osteoporosis and or broken bones from an early age with little force – see Osteogenesis Imperfecta (please check for this as well as hypermobility in all suspected child abuse cases TY)
THIS LIST IS NOT COMPREHENSIVE. It is just meant to give you an idea of some unusual signs to watch for. If someone is just hypermobile with generalized joint pain, some subluxations and or dislocations, easy bruising, some easy bleeding, IBS, headaches and myopia, but none of the above, they probably have hEDS. But please do proper thorough workup and differential diagnosis! Many cases of hastily diagnosed hEDS and HSD are turning out to be something else. And this may well affect treatment and prognosis, as well as epidemiology (statistics).
Diagnosing Hypermobile type EDS (hEDS) and Hypermobility Spectrum Disorders
Once you’ve ruled out all the more distinct rare forms of EDS and other HCTDs as described above, you are then left to diagnose the most common type of EDS by far known as hypermobile EDS or hEDS, clinically only via a thorough physical exam using these new criteria.
And if the patient is not bendy enough, or doesn’t pass those, then one of the forms of Hypermobility Spectrum Disorders should be assigned now as previously mentioned.
In other words, the Hypermobility Spectrum Disorders are a diagnosis of exclusion of ALL of the forms of EDS including the most common hEDS, and hypermobile type EDS (hEDS) is a diagnosis of exclusion of all of the other more rare, genetically-based forms of EDS as well as of all other similar Heritabler Connective Tissue Diseases (Marfans, Stickler’s, OI, Loeys-Dietz, etc.)
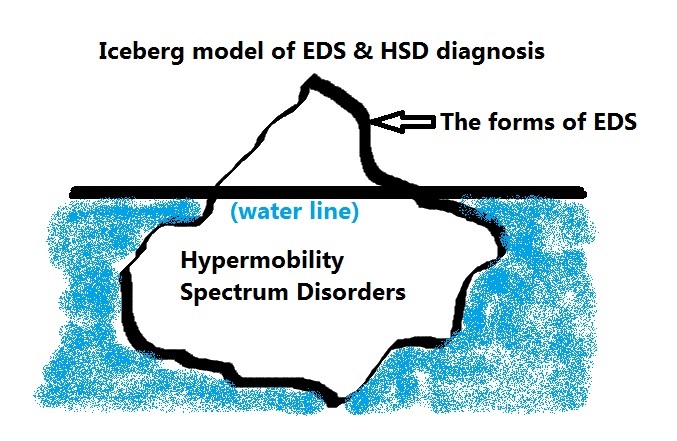
I like to think of the forms of EDS as the tip of the iceberg, easily visible above the water line and thus more easily diagnosed, and the new Hypermobility Spectrum Disorders are the vast majority that are less visible to the medical eye below the water line that we have just now recognized is there.
Diagnosing Vascular EDS (vEDS) in particular
Ehlers-Danlos of all types runs across all races/ethnicities. But thanks to more visible blue veins showing under their more “see-through” white skin, almost everyone with pale or lighter brown skin wonders at some point if they don’t have vascular EDS (vEDS, formerly Type IV), a dangerous rare form that lends to more shortened life spans than most from arterial or inner organ rupture. NB – vEDS can run in any race or ethnicity, not just white or Caucasian people.
Few of us will have vEDS, thankfully, but if you have enough other signs like a family history of vascular or internal organ ruptures under age 40 or aneurysms as shown here from the UW Collagen Diagnostic Lab who perform the testing for it, definitely rule it out:
“1. Possible diagnosis of EDS type IV: The vast majority of probands in families with this form of EDS are identified on the basis of a major complication either bowel perforation or vascular aneurysm or rupture. The International Ehlers-Danlos Foundation Advisory Board set the following guidelines for determination of the clinical diagnosis of EDS type IV. DNA-based testing is recommended for those who meet these guidelines. Note, however, that individuals with nonsense mutations of COL3A1 are less likely to have similar physical characteristics. The clinical diagnosis of EDS type IV is highly suspected when two major diagnostic criteria are present:
Major clinical diagnostic criteria:
- Intestinal rupture
- Arterial rupture
- Uterine rupture during pregnancy
- Family history of the vascular type of EDS
Minor diagnostic criteria alone are not sufficient to warrant the diagnosis unless identified in an individual with a major criteria.
- Thin, translucent skin (especially noticeable on the chest/abdomen)
- Easy bruising (spontaneous or with minimal trauma)
- Characteristic facial appearance (thin lips and philtrum, small chin, thin nose, large eyes)
- Acrogeria (an aged appearance to the extremities, particularly the hands)
- Hypermobility of small joints
- Tendon/muscle rupture
- Early-onset varicose veins
- Arteriovenous carotid-cavernous sinus fistula
- Pneumothorax/pneumohemothorax
- Chronic joint subluxations/dislocations
- Congenital dislocation of the hips
- Talipes equinovarus (clubfoot)
- Gingival recession”
“Proband” is genetic speak for “person presenting now” (I.e, the patient), vs their parents or children. I actually had vascular and kyphoscoliotic types ruled out based on a history of an ascending aortic aneurysm (or AAA) in my dad, skin and facial features, severe scoliosis in my aunt from birth, keratoconus, visible veins, and small joint involvement among other things. I was relieved to learn I was negative for both, and we settled on EDS-HT (now hEDS) for my diagnosis in 2012.
But that doesn’t mean I don’t still watch for issues, and I had a baseline echocardiogram done to rule out any aortic root dilation (common in us), or mitral valve issues, and watch for aneurysms.
Concluding notes
To be sure, there’s nothing very benign about hEDS aside from not dying directly. People with hEDS and the HSDs still end up mitigating plenty of serious issues, including severe dysautonomia, weak necks and spines lending to CNS issues, sciatica, spondylolysthesis (slipped vertebrae), Chiari malformation and more. (Including anaphylaxis from commonly comorbid Mast Cell issues.)
Further, although EDS types generally run “true” within a family, meaning if your parent has vEDS, you will get vEDS (if you do), it is possible to have more than one type of EDS coexisting in one person, even if it’s rare. I know one patient with both a rare form of EDS AND Marfan Syndrome, both confirmed with genetic testing. Talk about winning the genetic lottery!
But generally EDS will run true to type, meaning, if your contributing parent has classical EDS, you will inherit that form, and not kyphoscoliotic EDS, unless careful medical history or testing reveals another form from the other side of your family or you have a rare de novo mutation in your family. You probably can’t have more than two types though, as this may not allow for a fetus to be viable.
No, these conditions are no cake-walk, though yes, as I’m slowly but steadily proving along with many other hardy survivors that you can recover to some extent through dedicated nutritional & dietary work and tailored “zebra-friendly” (core-building) physical therapy that avoids injuring us further, and strengthening the bits we can.
Lastly, I’ll note that having EDS itself, as comprehensive as it is, does NOT preclude having other diseases, conditions or issues (aka comorbidities), simultaneously. As Hickam’s Dictum states: “patients can have as many diseases as they damn well please!” And I mean beyond the incredible list of ones I’ve already noted are fairly common in the community (thyroid issues, autism, depression, CVID – immune deficiencies, allergies, insomnia, chronic fatigue, fibromyalgia, etc.) You may have any number of other illnesses and genetic disorders in addition, also.
As someone else unknown to me brilliantly said, EDS is as individual as fingerprints, and we are all unique individuals in addition especially when you toss in environmental factors which influence our epigenetics, a whole other layer affecting genetic expression. So while we may share some common “themes” of dysautonomia, fatigue, pain and hypermobility, it will truly express absolutely uniquely within individuals even within the same family. This is known as variable expression. To borrow another phrase from the Autistic community: if you’ve seen one Ehlers-Danlos (or HSD) patient, you’ve seen one Ehlers-Danlos (or HSD) patient!
But as you know I feel strongly, every doctor has seen HSD if not also EDS patients whether they know it or not. Heck, we’re the frequent flyers in their offices by default! In fact, everyone has seen an HSD or EDS patient whether they know it or not, we’re that common IMHO. Hopefully the doctors will start spotting us a bit sooner with this information I’m sharing. (Nurses too!) I hope the above has helped to clarify how best to pursue a diagnosis once you do suspect it.
Thank you for this website!
Also, “patients can have as many diseases as they damn well please!”, is generally attributed to John Hickam, MD. It’s generally called Hickam’s dictum.
Hickam’s dictum is a counterargument to the use of Occam’s razor in the medical profession.[1] The principle is commonly stated: “Patients can have as many diseases as they damn well please”. The principle is attributed to John Hickam, MD. Hickam was a faculty member at Duke University in the 1950s, and was later chairman of medicine at Indiana University.[2]
Thank you! I’ve been trying to find the author of that great quote also for a while now, so thank you for sharing it! (I just asked in a Facebook group a couple weeks ago, but no dice.) Hickam’s Dictum – brilliant! And too true for us, also, eh. Ahh… my brain can rest now and stop searching for that answer finally.
I have HEDS and looking at the information here on VEDS, I have almost all the minor criteria but none of the major criteria. I suppose that’s good but I never worried about it at all and now I’m going hmmmmm. Good information though. Thank you.
A lot of us do, myself included. Definitely get tested for it if concerned and can get a doctor to order the test. It’s the one from UW Collagen Lab I believe I linked to in the article. Good luck!
Thank you for that excellent, and informative article. I am also very grateful to you for listing one of my books – but I have another – ‘A Guide to Living with Ehlers-Danlos Syndrome – Hypermobility Type – Bending without Breaking’ 2015, Singing Dragon Press/Amazon.
Keep writing!!
Thanks – I do know about your first book, but I think I thought your second one sort of supplanted it. I’ll defo add it back into my list if you’d like, no problem. It’s how I first learned about Bowen Technique working so well for us. (I love it, personally, just wish I could afford more.) Glad you found me!
Oh – I see now, you revised your first book! Okay, will relist, thanks.
Er- pale skin implies that you can only have VEDs if you are white. And this is not true.
I quite agree, it’s just that black people have not exhibited nearly the same anxiety levels as white people over their visible blue veins. I would suspect it based on family history of internal organ or vascular rupture, not appearances so much. (I show many Vascular features including visible veins but am negative based on actual genetic testing done by the UW lab.) It crosses all racial/ethnic bounds yes.
“The two most common types of EDS (hEDS and cEDS) have an autosomal dominant inheritance pattern.” False re hEDS. It can seem to have this pattern in some lineages, but for one (possibly majority) group, with the multiple-alpha-tryptase (TPSAB1) trait, only some get the disease – about a third. At a minimum, even if not environment but other genes make the difference, it’s not likely that the necessary collection of genes are dominant – it’s certainly not the case that we know that. Moreover, inheritance within the group that kicked off the study seems very mixed and complex.
This isn’t a small or exceptional group. The percentages of the population they’re talking about could pretty much encompass all HEDS cases, if my back of the envelope calculation is correct.
The study:
Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number
by Jonathan J Lyons et al
I will concede the point about not knowing for sure about hEDS being autosomal dominant. I may reword that – I’m going off what was pounded into me 5 years ago by others. I personally think it’s much higher actually, but have no scientific backup. However, I’ll gently disagree about this study possibly explaining a majority of hEDS cases on two fronts:
a) I was in it, and I do NOT have elevated tryptase, I promise (ask my doctor, we tried to elevate it, and no dice)
b) It really only explains a subset of the hEDS population. There are oodles of us like me with low tryptase, but still having hEDS (as best we can tell in the absence of a better dx, and yes, I tested for Vascular and Kypho by blood in 2012 based on phenotype) who are not yet explained.
But your point is well taken, and I may reword things a bit up there. We truly don’t really know yet until much larger studies of hEDS patients are done. I know for a fact (I’ve dialogued with Dr. Milner by email) that they only took patients with signs of MCAD and hEDS comorbid. I was put in touch with the study by Dr. Melody Carter who came up to me after I made a comment at the 2012 TMS Conference about the high rate of comorbidity of MCAD (all types) and EDS I was seeing in the groups. Most of the doctors denied this, but a couple grudgingly admitted to having a couple of known patients. I’m seeing this slowly change with time. That was October 2012…
Thanks for the feedback in any case! Rock on – Jan
Please bear with me as I get the above properly and fully re-written to reflect the major new nosology and criteria change for ALL forms of Ehlers-Danlos Sydnromes plus the new catgory of Hypermobility Spectrum Disorders for those who walk like a zebra, talk like a zebra but have ruled out all forms of EDS and other HDCTs. Sort of our new “catch-all” bin. Will probably cover many if not all/most with fibromyalgia I’m guessing.
Apologies and sympathy to all trying to parse the new criteria. Essentially, we now have 7 new forms of EDS reocgnized, for a total of 13 up from 6 before, and… all are now considered rare again, as they tightened the dx criteria for hEDS back up a bit.
But the new category of Hypermobiltiy Spectrum Disorders should cover all others whether mildly or more afflicted with any form of joint hypermobility and connective tissue issues after ruling out all other forms of both EDS and heritable connective tissue disorders (HCDTs) such as Marfan, OI etc.
Thanks for your patience! Jandroid March 19, 2017 (phew, typ-ing my fingers off!)
Thanks for all your work. Just curious, and confused. How do “older” or oldish people ever get diagnosed with hypermobile/type 3 if they come from small families and without children. I was recently dx at 49 with hypermoble (tho I think I may be classic), but I am not sure I would have been able to get a dx if you MUST have a diagnosed family member. My parents are in their 80’s and never had issues as bad as me, it was hard enough for me to be believed for dx. My brother has died. I have no cousins. Are those with small families just left out now? It looks to be a required criteria. I don’t understand. Sorry if I am not yet using the correct nosology. Just was curious for another opinion.
Best to you!
I was an “older” patient when I finally got diagnosed, but alas, only because I went suddenly from walking to wheelchair in 3 weeks in early 2012 just in time to turn 45 ~! Ha – now my doctor believes me about all my pain. 😉 (I was “just a woman” or “just depressed” before, lol. If only!) Anyway, my parents were gone, and my older sister wasn’t speaking to me. I gathered as much ifno as I could remember and solicited from my living aunties and a cousin and filled out a spreadsheet with their names across the top, and symptoms down the left side. (You tick off the boxes under each name as fits – it becomes really clear who is most “symptomatic” and likely had it too that way.) It very clearly ran on my dada’s side for sure, though I now suspect my late mom in 20/20 hindsight too. (May explain my “severity” among other epigenetic drivers.)
Anyway, family history is not required – just helpful. It is only 1 of 2 things required to pass criteria 2 for hEDS now. If you pass the other 2 you’re still okay even without a diagnosed family member. It’s just beneficial to have one. Not required. Some info is here:
http://ehlers-danlos.com/eds-diagnostics/
And more here:
http://ehlers-danlos.com/2017-eds-international-classification/
Good luck, I hope that helps.
Here’s the actual hEDS criterai now. To clarify: you can pass Criterion 2 if you pass either Features A and C, or A and B, or B and C. (You don’t have to pass all 3, A, B AND C. Just any two of them.) You do not HAVE to pass B – which is the family member. It’s just helpful.
E.g., if you don’t pass A, but you DO pass B and C, then you pass Criteria 2. So it would be helpful then.
Alternatively, if you don’t pass C but you DO pass A and B it helps.
If you pass A and C but NOT B (because you can’t find your family, they are not diagnosed yet/at all/ or not speaking to you like mine, or are adopted E.g.), you still pass criterion 2. I hope that makes sense.
I.e, you only need 2 out of the 3 “Features” (A, B or C) in Criterion 2. Yes, a wee confusing! x http://ehlers-danlos.com/wp-content/uploads/hEDSvHSD.pdf
Three short and related questions:
(1) Because of the new stringency on diagnosing hEDS, will healthcare insurance companies still recognize the more generic HEDS diagnosis (so we can take tax deductions for yoga, PT, etc.) OR is a specific ED diagnosis now required?
(2) If a generic HEDS diagnosis no longer medically valid for insurance purposes, will healthcare insurance companies recognize a specific HSSD diagnosis (so we can take tax deductions for yoga, PT, etc.)
(3) Of all the possible HSD diagnoses, which one is the most likely place you’d slot Osgood-Schlatter?
I’m newly researching EDS and truly appreciate the work you’ve put into OhTwist. My best to you.
Aw thank you so much. To answer your short but packed questions:
“(1) Because of the new stringency on diagnosing hEDS, will healthcare insurance companies still recognize the more generic HEDS diagnosis (so we can take tax deductions for yoga, PT, etc.) OR is a specific ED diagnosis now required?”
Great Q, I honestly don’t know. I’m betting it may vary from company to company, depending on their level of awareness and education around the condition. I would try asking THe Ehlers-Danlos Society via info@ehlers-danlos.com
“(2) If a generic HEDS diagnosis no longer medically valid for insurance purposes, will healthcare insurance companies recognize a specific HSSD diagnosis (so we can take tax deductions for yoga, PT, etc.)”
Another really great Q! And I wish I knew, and I dearly hope they do, but hard to say. If I had to guess, they may dismiss at least the asymptomatic forms of HSD since it’s technically not a problem for them – yet. (There are 3 asymptomatic categories and the same three plus one more symptomatic categories of HSD for a total of 7.) As shown on the last page here: https://ehlers-danlos.com/wp-content/uploads/hEDSvHSD.pdf
“(3) Of all the possible HSD diagnoses, which one is the most likely place you’d slot Osgood-Schlatter?”
Another great Q! I’m not sure it might not be found across several types/forms frankly, as I think it has to do with speed of your bony plate growth no? But I honestly don’t know. Again I’d try the EDS here: info@ehlers-danlos.com.
Sorry I’m not much help, but these are excellent Qs for all of us! Good luck!
Thank you for this. Tough to zero in on a moving target! As you suggest, hopefully all our queries trigger a new way of looking at these issues.
Okay, I think (think!) I may have finally gotten this sucker sufficiently updated to not confuse the heck out of any new visitors. Let me know if you see any contradictions or inconsistencies, thanks! Jan – April 28, 2017.
Im trying to find a genetics specialist. To see if i have EDS . My brother and 2 out of his 3 children also have it…im having so many issues and i truely believe this cld be the answer to my problems …CLD SOMEONE PLEASE HELP ME WITH SOME NAMES AND #’s
Hi Amie, I’m glad you’re looking for a geneticist to help diagnose your family since you suspect this condition. First I need to know what part of the world you’re in to start to answer that. But since I don’t, I’ll guess you may be in the US, and if so, try finding a local or nearby support group to you here:
https://www.chronicpainpartners.com/
And also try these guys: http://ehlers-danlos.com
And if you’re not in the US, check out this page here:
https://www.rareconnect.org/en/community/ehlers-danlos-syndrome/members
I hope that helps. I strongly encourage joining the International Ehlers-Danlos Society (second link above) for support on your journey. And, you can find lots of groups and support on Facebook too if you happen to be on there. As well as also here on Inspire:
https://www.inspire.com/groups/ehlers-danlos-syndromes-and-related-disorders/
Take a deep breath, you will get some answers. Think marathon, not sprint, and start gathering that family history. Glad I could help. Good luck!
Thank you for this wonderful post with so much info! I finally have a referral to genetics for EDS. I am not sure if I will be considered bendy enough, as I have a lot of hypermobility in my hands and in my neck/spine, but only just barely obvious on my left side. Constant subluxations but little visible hyperextension in large joints. I currently have a diagnosis of fibro and joint hypermobility, but it’s never covered all of my many (T)issues.
One slight correction I will add is that the “autism community” and “autistic community” are diametrically seperate entities and the quote should be attributed to the latter. The first represents parents, family, and professionals, and is for the most part made up of the medical model of autism, whereas the latter represents the autistic community ourselves.
Remember that if you are at all bendy, even if not much, you can still possibly qualify for a diagnosis of the new category of Hypermobility Spectrum Disorders. This is the new diagnosis of exclusion after all other more closely matching forms of CTDS and EDS are ruled out for you. But you still have several signs. It’s just so newly recognized that few doctors have heard of it yet. I urge using the EDS Toolkit here for help: http://rcgp.org.uk/eds.
And thank you for that clarification of the terminology between “autism community” and “autistic community”! I’m still so new to the social model and the autistic community that I’m still prone to slipping up. Thanks for your patient education! I’ll slowly get there… but I’d like to get it right so as to bring more with me.
Hi, Please help me to understand how diagnosis for any EDS form will effect medical insurance.
I’m so sorry, but that is one area I really don’t feel able to help with. Not only do I think it may differ, depending on your type of insurance, but I don’t think it should matter, frankly. It’s more a matter of what doctors you have access to, and their views of EDS etc. regardless of your insurance, IMHO.
Where I have heard it mattering, is with life insurance – it seems to be seen as a black mark against us there.
Sorry not to be more help! Anyone else? Please chime in if you can help answer this thanks!