Regarding HSD vs hEDS
This page and topic are currently somewhat controversial, and have been ever since the inception (invention) of the Hypermobility Spectrum Disorders in 2017 by those claiming the power to decide these things.
These powers chose a stereotyped and limited subset of criteria and signs to come up with the diagnostic criteria for hypermobile type EDS (hEDS) designed to support research into the underlying gene or genes, rather than to aid patient care and quality of life. hEDS is considered the most common type of EDS by far, but prevalence rates are disputed as more robust epidemiology is lacking.
The current ( as of October 2025 still) 2017 hEDS diagnostic criteria skew toward thinner white bodies with more apparent Marfanoid habitus. They are also biased toward people who do not have additional autoimmune diseases (which we frequently do), as this added condition is then literally held against the patient, dismissing their recurrent joint dislocations or frank instability (Feature C, Criterion 2) in Criterion 3. Plus, many will be stiffer, and thus have a lower Beighton score due to arthritis or other autoimmune diseases. Thus this is also held against them, making it even harder to be diagnosed with hEDS if you have acquired a secondary autoimmune disease as many have. (Often diagnosed before their hEDS/EDS/HSD.)
And they further discriminate by requiring patients to have biological family available to help make the diagnosis in those with co-occurring autoimmune diseases. Not everyone has access to their families of origin, either because they were adopted, abused or abandoned, and some may never. Nor are all biological families cooperative. And many of our kin are actually homeless, lacking any access to proper medical care.
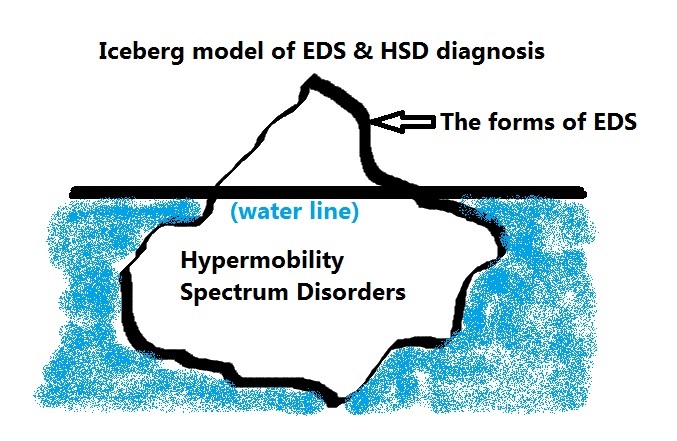
There really is no clean, clear distinction between hEDS and HSD yet. ETA 9/15/24: And the recent finding of a potential biomarker common to BOTH hEDS and HSD patients (a small finbronectin fragment in blood) announced September 3, 2024 may serve to obliterate this distinction soon. HSD has truly just been the newest “catch-all bin”, or diagnosis of exclusion – to hEDS. Which itself was a prior catch-all bin or diagnosis of exclusion of all other forms of Ehlers-Danlos syndromes and similar Connective Tissue Diseases/Disorders (CTDs) like Marfan Syndrome, Stickler Syndrome, Osteogenesis Imperfecta and Loeys-Dietz Syndrome. These all share similar issues of hypermobility and weak connective tissues and many attendant issues (sequalae, or knock-on effects of weak tissues), but from different underlying molecular causes than ascribed to the forms of EDS.
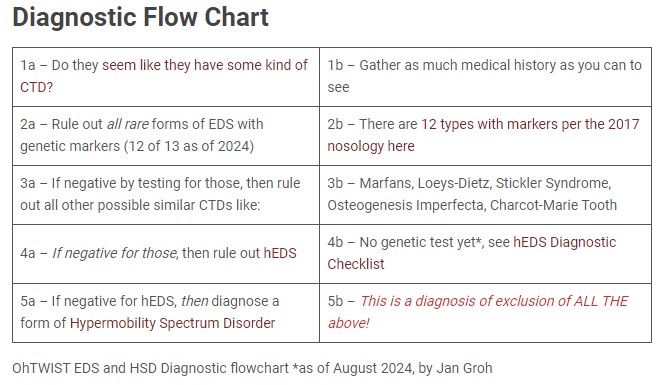
Image ID: a 2×5 table titled “Diagnostic Flowchart” reading:
- 1a – Do they seem like they have some kind of CTD? 1b- Gather as much medical history as you can to see
- 2a – Rule out all rare forms of EDS with genetic markers (12 of 13 as of 2024) 2b – There are 12 types with markers per the 2017 nosology here
- 3a – If negative by testing for those, then rule out all other possible similar CTDs like: 3b – Marfans, Loeys-Dietz, Stickler Syndrome, Osteogenesis Imperfecta, Charcot-Marie Tooth
- 4a – If negative for those, then rule out hEDS 4b – No genetic test yet*, see hEDS Diagnostic Checklist
- 5a – If negative for hEDS, then diagnose a form of Hypermobility Spectrum Disorder 5b – This is a diagnosis of exclusion of ALL THE above!
OhTWIST EDS and HSD Diagnostic flowchart * – created August 2024 by Jan Groh. Kept in October 2025.
In other words, you are supposed to thoroughly rule out not just all other rare forms of EDS (of which there are now 12 official per the 2017 nosology), but also all other similar appearing CTDs (as mentioned above) in order to diagnose hypermobile type EDS. (Not always done.)
But if you run the hypermobile EDS (hEDS) criteria checklist, and someone doesn’t quite pass despite having many signs and suffering (else why are they at a doctor’s office), and you have thoroughly ruled out all other possibilities mentioned above, and they still have pain and issues with their tissues and signs not caused by an autoimmune disease, THEN, you can diagnose someone with a form of the Hypermobility Spectrum Disorders as a final resort. (Phew, piece of cake – not.)
There are no actual criteria for the HSDs yet though this is under discussion again in 2023. Nor is there an ICD-10 code yet. It is my dearest hope they get folded back into the hEDS bin, and the biomarker and or multiple genes (MIA3, Kallikrein – see below) assigned. (THANK YOU, SCIENTISTS!)
The following are articles that either help describe, diagnose, recognize or define either hypermobile type EDS (hEDS) or the Hypermobility Spectrum Disorders. As well as describe how they differ, or if they really do. And will include the latest research into hEDS and or HSD.
See also The RCCX Theory (yes, technically a hypothesis yet) on a possible genetic complex behind hEDS/HSD. (I gave this its own page as its such a complex topic that has no bench research backing it yet either.) Updated October 25, 2025
Hypotheses on causes of hEDS or HSD and how they may differ or not
- Proteomic discoveries in hypermobile Ehlers–Danlos syndrome reveal insights into disease pathophysiology October 2025 Griggs, Daylor, Petrucci et al
- Mitochondrial Dysfunction and Its Potential Molecular Interplay in Hypermobile Ehlers–Danlos Syndrome February 2025 Shirvani, Shirvani and Holick
- Hypermobile Ehlers–Danlos Syndrome: Diagnostic Challenges and the Role of Genetic Testing April 2025 Forghani, See and McGonigle
- Bridging the Diagnostic Gap for Hypermobile Ehlers-Danlos Syndrome and Hypermobility Spectrum Disorders: Evidence of a Common Extracellular Matrix Fragmentation Pattern in Patient Plasma as a Potential Biomarker September 2024 Ritelli, Chiarelli, Cinquina et al Am J of Am Med Gen
- Variants in the Kallikrein Gene Family and Hypermobile Ehlers-Danlos Syndrome Preprint released June 2024 by Gensemer, Beck, Guo et al (news via CPP here)
- Phenotypic Clusters and Multimorbidity in Hypermobile Ehlers-Danlos Syndrome 2024 Petrucci, Barclay, Gensemer et al
- A novel mutation in collagen transport protein, MIA3 gene, detected in a patient with clinical symptoms of Ehlers–Danlos hypermobile syndrome Junkiert-Czarnecka, Pilarska-Deltow, Bak et al January 2023
- Folate-Dependent Hypermobility: Researchers at Tulane’s EDS Clinic Look Into New Possible Mechanism For Hypermobile EDS via EDS Awareness May 2023 (Here is the Tulane paper itself.)
- Genomic Characterization by Whole-Exome Sequencing of Hypermobility Spectrum Disorder 2022 Alanis-Funes, Lira-Albarran, Hernandez-Perez et al
- Are patients with hypermobile Ehlers–Danlos syndrome or hypermobility spectrum disorder so different? 2021 Aubry-Rozier, Schwitzguebel, Valerio et al
- The Role of Cell Adhesion and Cytoskeleton Dynamics in the Pathogenesis of the Ehlers-Danlos Syndromes and Hypermobility Spectrum Disorders 2021 Malek, Koster
- Hypermobile Ehlers‐Danlos syndromes: Complex phenotypes, challenging diagnoses, and poorly understood causes 2020 Cortney Gensemer Randall Burks Steven Kautz Daniel P. Judge Mark Lavallee Russell A. Norris
- RNA-Seq of Dermal Fibroblasts from Patients with Hypermobile Ehlers–Danlos Syndrome and Hypermobility Spectrum Disorders Supports Their Categorization as a Single Entity with Involvement of Extracellular Matrix Degrading and Proinflammatory Pathomechanisms December 2022 Ritelli, Chiarelli, Cinquina et al Dec 2022
- Mechanobiology in the Comorbidities of Ehlers Danlos Syndrome April 25, 2022 Shaina P.Royer and Sangyoon J. Han
- Some cases of hypermobile Ehlers-Danlos syndrome may be rooted in mast cell activations syndrome 2021 Afrin, Lawrence, B MD
- MUSC researchers announce gene mutation study linked to EDS 2021
- Matrix Metalloproteinases Inhibition by Doxycycline Rescues Extracellular Matrix Organization and Partly Reverts Myofibroblast Differentiation in Hypermobile Ehlers-Danlos Syndrome Dermal Fibroblasts: A Potential Therapeutic Target? 2021 Chiarelli, Zoppi, Venturini et al
- Fibromyalgia – an unrecognized Ehlers-Danlos Syndrome Hypermobile Type? (in French) 2013
- Dermal fibroblast-to-myofibroblast transition sustained by αvß3 integrin-ILK-Snail1/Slug signaling is a common feature for hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders 2018 Nicoletta Zoppi, Nicola Chiarelli, Silvia Binetti, Marco Ritelli, Marina Colombi
- Human Mast Cell beta-Tryptase Is a Gelatinase 2003 Fajardo and Pejler
Discussions of Prevalence of hEDS or HSD
- Diagnosed prevalence of Ehlers-Danlos syndrome and hypermobility spectrum disorder in Wales, UK: a national electronic cohort study and case–control comparison 2019 Joanne C Demmler, Mark D Atkinson, Emma J Reinhold, Ernest Choy, Ronan A Lyons, Sinead T Brophy
- Hypermobile Ehlers–Danlos syndrome (a.k.a. Ehlers–Danlos syndrome Type III and Ehlers–Danlos syndrome hypermobility type): Clinical description and natural history (pages 48–69) 2017 Brad Tinkle, Marco Castori, Britta Berglund, Helen Cohen, Rodney Grahame, Hanadi Kazkaz and Howard Levy
- Childbearing with Hypermobile Ehlers–Danlos Syndrome and Hypermobility Spectrum Disorders: A Large International Survey of Outcomes and Complications 2023 Pearce, Bell, Pezaro, Reinhold
- Hope for Hypermobility: Part 1—An Integrative Approach to Treating Symptomatic Joint Hypermobility 2023 Daylor, Victoria BFA; Gensemer, Cortney PhD; Norris, Russell A. PhD; Bluestein, Linda MD
- Hope for Hypermobility: Part 2—An Integrative Approach to Treating Symptomatic Joint Hypermobility 2023 Daylor, Victoria BFA; Gensemer, Cortney PhD; Norris, Russell A. PhD; Bluestein, Linda MD
- Ehlers-Danlos Syndrome Hypermobility type: A Much Neglected Multi-Systemic Disorder Gazit, Jacob & Grahame 2016 Rambam
- Ehlers-Danlos Syndrome (EDS), an Hereditary, Frequent and Disabling Disease, Victim of Iatrogenia due to Widespread Ignorance Harmonet 2015
- The Lack of Clinical Distinction Between the Hypermobility Type of Ehlers-Danlos Syndrome and the Joint Hypermobility Syndrome (a.k.a. Hypermobility Syndrome) Tinkle, Bird et al American Journal of Medical Genetics Part A 149A(11):2368-70 · November 2009 (Also available as an article on p 17 of Autumn 2010 EDNF Loose Connections )