About EDS
May 2, 2017: The new Ehlers-Danlos Syndrome nosology (classification system) will now refer to all of the forms EDS as a collection, so in the plural, as “the Ehlers-Danlos Syndromes” (with an “s”) now.
They are among the most common forms of Heritable Connective Tissue Disorders (or HCTDs), and share overlapping signs with some others, including Marfan Syndrome, Sticklers Syndrome and Osteogenesis Imperfecta among others. Heritable means you were born with the disease or trait at birth, having gotten it from one or both parents.
Connective tissue is what holds the body together, including our skin, tendons, ligaments, fascia, blood vessels and more. Most is made from collagen, which runs through 80% of the body. So faulty collagen formation can wreak a lot of havoc, lending to so many issues.
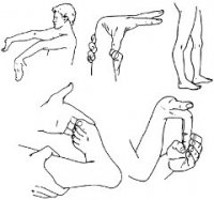
Hypermobility by itself is just a trait, like red hair color and is generally though to be benign by itself. The Ehlers-Danlos Syndromes all involve hypermobility plus additional issues that drive you to see a doctor, not least of which includes wide-spread pain. Yes, many may be getting under or mis-diagnosed with Fibromyalgia yet, in my unprofessional opinion. It runs across all ages and races and skin types though most photos are of Caucasians currently. (Hopefully this will change with time.)
Hypermobility Spectrum Disorders (HSDs)
Furthermore, there is a BRAND NEW diagnostic category since March 2017 available also called the “Hypermobiltiy Spectrum Disorders” or HSDs for those who are a wee too sub-clinical to meet the newly tightened hypermobile type EDS or hEDS criteria, appear negative for anything else, but are still plenty symptomatic. Trust me, they feel our pain, and are hoping to get it treated. That’s the goal of this new category. That said, it remains to be seen if doctors take this as seriously as we do. The intention is that they sure should. The international working group just wanted a tighter phenotype for hEDS to help speed up research is all. Think of the HSDs as the less clinically visible bulk of the iceberg below the water line, as in this image:
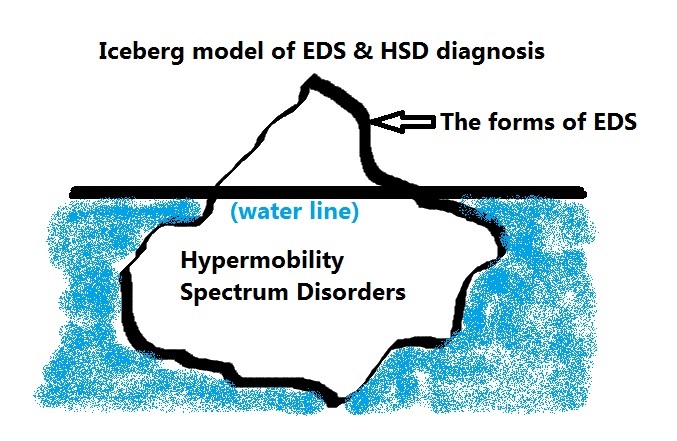
The true prevalence of the HSDs is unknown yet, having only recently been recognized. But it is believed they are much more common than the forms of the Ehlers-Danlos syndromes that are more clinically visible and better known to date. (It is my goal to help change this with this site.) It is possible many with fibromyalgia may really have a form of HSD or hEDS.
Most forms of EDS run “true” in a family, meaning if your mother had vascular EDS (vEDS), you will inherit vEDS if any. But it does NOT preclude having more than one form, as you do have two parents. Nor does it preclude having other diseases and disorders on top of or in addition to these painful syndromes a la Hickam’s Dictum. (Interestingly I personally have found the hypermobile form and/or what we now call the HSDs to often come with what I call a “Chronic Constellation” of conditions that are yet to be scientifically correlated or connected, so I present this only as a very unscientific observation as yet.)
The same can’t yet be said of the HSDs – they are too new yet. I personally think most with hypermobility, symptomatic or not to likely be related to someone with a form of EDS, but again this is pure speculation and remains to be seen. Hopefully molecular testing will catch up quickly and we can all stop wondering and have more certainty, instead of hoping for doctors to take our complaints seriously in the absence of easy clinical markers.
All of the forms of Ehlers-Danlos Syndrome and the new HSDs involve faulty collagen formation from a variety of causes, including genetic collagen defects, or other genetic defects causing insufficient or poorly aligned collagen formation in the extra-cellular matrix.
Collagen
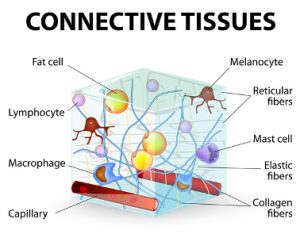 Collagen runs throughout the entire body, thus all systems are ultimately affected to varying degrees, and the most obvious and easy to spot to date have been the skin and joints. In the past, only cases with stretchy skin were diagnosed. It is now recognized that not all with some forms of EDS have stretchy skin. Further, not all have equally hypermobile or “bendy” (flexible) joints, whence the new categories of Hypermobility Spectrum Disorders have been added, and different “flavors” of hypermobility described:
Collagen runs throughout the entire body, thus all systems are ultimately affected to varying degrees, and the most obvious and easy to spot to date have been the skin and joints. In the past, only cases with stretchy skin were diagnosed. It is now recognized that not all with some forms of EDS have stretchy skin. Further, not all have equally hypermobile or “bendy” (flexible) joints, whence the new categories of Hypermobility Spectrum Disorders have been added, and different “flavors” of hypermobility described:
- Generalized – you show generalized (body-wide) hypermobility, usually passing the age and sex specific Beighton scale or score for the diagnostic criteria used
- Peripheral – you generally show hypermobility primarily in the hands and feet*
- Localized – you may only show hypermobility (or flexibility/laxity/instability) in just one or two joints, e.g. just your knees, or just an ankle or one bad hip or shoulder.
* This can be a flag to look for more signs of the vascular type of EDS including a history of internal organ, bowel or vascular weakness or rupture in the family among many other signs, most notably extremely thin see-through “aged” looking skin (acrogeria) even in young adults, and aneurysms. By all means rule this out if suspected!
The forms of EDS and also the HSDs can lend to weak tendons, ligaments, prolapsed organs, weak blood vessels and valves, stretchy innards that may tear easily (like thin tire walls right?), herniations, subluxations and the most obvious full dislocations among many other things which can even be common for some.
Myopia, easy bruising, fallen arches and flaccid veins are other common signs, but there are many more I’ve shared here to help people suspect the hypermobile form or one of the Hypermobility Spectrum Disorders for which there is as yet no known genetic marker, and so can only be diagnosed clinically by a savvy doctor.
Hypermobile EDS or hEDS, still has no known genetic marker underlying it yet, and remains the most common form, even if the Ehlers-Danlos Society now prefer to call it rare again. The other 13 types (totaling 14 now as of April 2018) are considered truly rare, some extremely so. But hEDS still requires a clinical diagnoses based on these new criteria.
That said, any doctor can technically diagnose this form who is willing to run the criteria. I would urge referring someone on to a geneticist if you suspect one of the more rare forms, as they are better versed in the signs, and gathering thorough family histories whenever possible to this end.
New HSD Diagnosis
BJHS, JHS and HMS and the Brighton (with an “r”) Diagnostic Criteria are all going away or being rolled up into the new HSD diagnosis, but the Beighton 9 pt hypermobility scale remains. At least there will be a little less confusion between those two names going forward accordingly. (Only Beighton with an “e” remains to my dismay.) I’m unsure how they will handle existing diagnoses of HMS, JHS and BJHS. I would contact The EDS at info@oreds.org to ask, or inquire with The Hypermobility Syndromes Association of the UK.
The Ehlers-Danlos Society just published an FAQ about the new EDS Nosology in March 2017 to help further explain the changes in views and terminology for everyone for the first time in 20 years. They are continuing to try to use descriptive names, even for the new extremely rare types as they’ve been trying to get us to do for years.

But you may occasionally see reference to the outdated “six main types” using numbers still, as shown here:
- Hypermobile Type or HEDS (formerly EDS Type III) – quite common, not well defined yet, many diagnosed with “fibromyalgia” instead, no single tissue or blood marker identified yet (see below for more on how to diagnose it)
- Classical Type or CEDS (formerly Types I and II) – semi-rare (1 in 5,000 -10,000), markers defined
- Vascular Type or VEDS (formerly Type IV) – rare (1 in 20,000) and life-threatening from internal organ or vascular ruptures
- Kyphoscoliotic Type or KEDS (formerly Type VI, also called Ocular-Scoliotic by the UW) – quite rare
- Arthrochalasiac Type or AEDS (formerly Types VIIA and VIIB) – quite rare (2-300 people worldwide approximately)
- Dermatosparaxis Type or DEDS (formerly Type VIIC) – extremely rare (<100 people worldwide?)
PLEASE FIND THE 8 ADDITIONAL NEW TYPES of EDS here now.
Lots of websites (many on my EDS resources page) still need to be updated, and so please take all with a grain of salt and bear with them patiently. Not all are being actively maintained any more, and many are run by fellow afflicted patients, not all of whom are keeping up as well or can. This is also the FIRST CHANGE IN 20 YEARS! So it’s a lot to take in and update after such a long run. But I expect more changes will come in the near future if they haven’t already too.
PLEASE REFRAIN FROM REFERRING TO THE OLD NUMBERING SYSTEM whenever possible to avoid confusion, and help move everyone onto the same diagnostic page, thanks! Yes, some of you were diagnosed back when those were used. Hopefully your medical systems will start to catch up. You can now point them to this FAQ explaining this if not!
I will continue trying to flesh this description out a bit more in time, but will leave it at that for now. I hope this has given you an idea of what constitutes the now plural Ehlers-Danlos Syndromes.
References:
US National Library of Medicine – Ehlers-Danlos Syndrome (Connective Tissue)