Tips for Doctors
 I’m not a doctor, but… I AM a very well spoken, studied and observant hypermobile Ehlers-Danlos Syndrome patient with a lot of experience seeing doctors and reading about other people’s experiences of same too. And like many with hEDS, I also have MCAS and had mild POTS (a form of dysautonomia), which we find commonly co-morbid, and sometimes even more show-stopping at my worst.
I’m not a doctor, but… I AM a very well spoken, studied and observant hypermobile Ehlers-Danlos Syndrome patient with a lot of experience seeing doctors and reading about other people’s experiences of same too. And like many with hEDS, I also have MCAS and had mild POTS (a form of dysautonomia), which we find commonly co-morbid, and sometimes even more show-stopping at my worst.
In a hurry to diagnose someone? Check out the EDS Toolkit for Doctors now here:
https://gptoolkit.ehlers-danlos.org
I will summarize some of the key features and considerations we complain about the most in our support groups below, to help our visits go more smoothly. I’ve been observing upwards of 30,000 patients (albeit socially) daily online through my various online support groups – and we love to complain!
So ironically I’m probably privy to more patient “complaints” than any doctor living who isn’t also in the support groups, as we’re all willing and able to share more in our online groups than we can in your offices, unfortunately for both of us. So I’m “seeing” lots of patients daily, albeit just “socially”.
So I’ve read a lot, and will summarize the biggest issues & most common complaints and misunderstandings that I believe will help the most here. Thank you for your consideration. It will save BOTH of us a lot of time, grief and money!
NB There is a BRAND NEW DIAGNOSTIC CATEGORY in addition to hEDS since 2017 called:
Hypermobility Spectrum Disorders
Think hEDS-adjacent, but with just as much trouble for some if not more, and much more common than EDS.
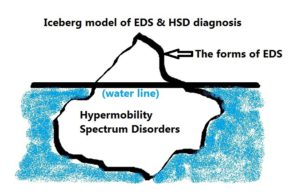
TL;DR: See Resources For Doctors here now
Most Common Issues and Chief Complaints
1) Assume we have vascular Ehlers-Danlos Syndrome when we present in the Emergency Department (A&E, ER or ED) until and unless proven or told otherwise. Better safe than sorry! This per The Ehlers-Danlos Society (formerly the EDNF), not just me.
2) The Ehlers-Danlos Syndromes (and now also the HSDs) are as individual as fingerprints, and no two patients will present alike, even within the same family and type. Not all will be equally bendy, or even visibly so. Nor will have the same pain levels, or tolerance, nor reactions. (See following points for more on these issues.) Yes, confusing.
Why? Who knows. I’m guessing it’s because some may have double alleles, vs single alleles, and epigenetics and several common comorbidities likely lend a role in this variation also, especially Sensory Processing Disorder which can swing hypo or hyper. It really presents on more of a spectrum or continuum much like autism. And, no kidding, Autism Spectrum Disorders are also very commonly comorbid.
3) We have paradoxic medication reactions – expect ANYTHING! And I truly mean anything. Not only do we NOT get much pain relief from the “caines” (Novocaine, Lidocaine, etc.) and often need way more than the average patient because we process them out too quickly due to our baseline hyperadrenergia and various CYP450 defects, but we may react badly to any of the opioids, and some may do nothing at all for us.
Yes, frustrating for both of us, I promise! We don’t like it either. Be prepared to treat anaphylaxis of any level at any time, and please BELIEVE us when we tell you we are feeling funny or reacting (often making us seem anxious, which is common in early mild anaphylaxis), or not getting sufficient pain relief.
{kind=link}
We are not drug seeking, I promise! We are all in way too much pain to become addicted – we almost never get high from opioids – we’re too afflicted. And because this condition is systemic, and lifelong/ongoing, we can be in nearly constant, relentless pain or re-injury at all times depending on our level of progression with this syndrome. (I was pretty mild and clearly sub-clinical up until I “fell apart” head to toe in 2012 at age 45 myself e.g.) It’s just not visible to you, alas. (See next.)
4) The causes of our pain rarely show on any scans – x-rays, MRI’s, CT’s, etc. except in the grossest cases, but our pain is still very very real. (We’ve usually been in serious agony for a while before something finally becomes visible, if ever.) We can feel loads of pain from micro-tears, strains, partial subluxations (too small to detect but EXTREMELY painful for some of us), and of course full dislocations and the resulting neuralgias, neuropathies, radiculopathies and the very common comorbid (or misdiagnosed) fibromyalgia. Small fiber neuropathy and mild demyelination are relatively common in the groups.
Many of us seem prone to CRPS and even arachnoiditis (along with springing CSF leaks), sadly too I’ve noticed. Please believe us when we say we are still hurting, for what seems like no apparent reason.
That said, a handful of lucky EDS patients can even pop their joints in and out at will, almost like a game – they shouldn’t do this, however – at some point the joint will stop going back in, with no warning! Others of us are in agony at the slightest pressure.
Also, do NOT force us to show how far we can bend – I’ve seen more patients be permanently injured this way than I can count! Yet they are not believed, sigh. (Thanks for reading and getting up to date here! Remember, first do no harm?) And yes, it can hurt, very very badly. I personally still injure in my sleep, and hurt from the minute I wake up until I fall asleep again. Further, contrary to what you’ve been told in med school:
5) Not all of us are very flexible or “bendy”, though we are still hypermobile. (Yes, there’s a difference.) Those who are less bendy may now be diagnosed with the new (as of 2017) category called Hypermobility Spectrum Disorders that is now used for such “stiff” patients as I am now. I used to be very bendy as a child but stiffened greatly with age. And some, like Dr. Jaime Bravo himself in Chile never are, he told me so in an email. Please read up on it! It’s brand new, and likely not rare! (It may well explain many “fibro” patients in fact.) Please read about how to properly diagnose us here.
6) Anaphylaxis comes in grades, and not all is throat-closing! Further, while we may not present at the traditionally understood and accepted level you have come to understand is full anaphylaxis, or not as quickly as you expect, we can and may tip over into that level (the highest grade on whatever chart you use) at any time just from stress, vibration, hot or cold, bright lights, sounds, foods, scents, medications etc.
{kind=link}
And… thanks to the crazy-making commonly co-occurring but still poorly known and rarely recognized Mast Cell Activation Disorders (MCAD) of any type (either Mastocytosis or the new and barely known MCAS or HaTS since 2016), we can react with anaphylaxis to almost ANY SUBSTANCE!
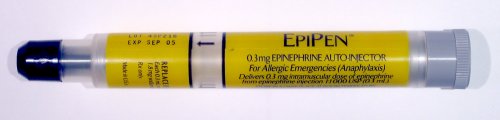
Yes, including non-protein substances like perfumes, dyes, fluoride, chlorine and yes, in one case I know, even the glucose in an IV infusion, no kidding! Yes, this is truly crazy-making and hard to believe, I understand. Seriously expect ANYTHING with us, as I stated at the top. I now believe a lot of MCAD is currently diagnosed as Multiple Chemical Sensitivity in fibromyalgia patients, but this is just my unverified theory again.
And, per The Mast Cell Diseases Society, patients on beta-blockers need a shot of glucagon along with any shot of Epi. (Patients who are NOT on beta-blockers do NOT need a shot of glucagon along with their Epi). You can consult The Medical Advisory Board of TMS for additional help in a crisis if needed: http://tmsforacure.org.
7) Anaphylaxis often presents like an anxiety attack, but it is not! Anaphylaxis can cause feelings of deep anxiety and impending doom in patients, and the accompanying signs and symptoms of lower-level or slowly building anaphylaxis will be mistaken for anxiety often until it’s too late. A low level reaction can suddenly “tip over” to full on “traditional” Epi-pen warranting Ana at any time.
Please trust us when we present in your ER saying “I’m having a reaction, can I please have an Epi shot?” or ask for IV saline solution or Benadryl. At the very least, please be willing to monitor us until the episode has passed, with Epi handy. While we do all try to carry and use our own Epi pens properly as much as possible (and usually not often enough), sometimes circumstances conspire to leave us short-handed.
Further, we can react strongly (from hyperadrenergia) to the Epi shots, and so we like to be in an ER where our symptoms can be better managed whenever possible. And, many don’t even realize they are having anaphylaxis yet, or know why they feel sick and anxious if they haven’t become acquainted with EDS and MCAD yet.
You or your nurses may be the ones to help ferret this out! I’m now increasingly convinced that a majority of cases that present to the ER with some cardiac involvement or angina and get diagnosed as “just anxiety” were low level anaphylactoid events that just aren’t being recognized as such yet, but that’s just my opinion.
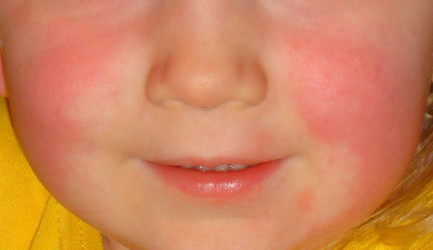
Common signs of MCAD reactions include easy flushing red of the mantle (upper neck, face and ears), hives, dermatographia during reactions, heat and other “rashes”, itching, terrible headache from IH, frequent urination, hydrocephalus, sudden nausea or diarrhea and sudden mood swings, irritability, emotional outbursts and temper flares. (We call this the “histameanies” btw.)
8) We often just need to rehydrate with an IV saline infusion due to our POTS or MCAD. We are prone to low blood volume from several causes including hypovolemia (stretchy bladders lend to urinating more than we take in), third-spacing from MCAD causing blood vessels to dilate and our plasma to flow out into our tissues (shown as bloating or angio-edema almost anywhere, including the lips and eyes). Again, it helps if you just believe us on this one -it’ll be one of the cheapest “fixes” you’ll ever give us.
We also often have electrolyte imbalances from unknown reasons, but especially salt and potassium. Not all patients know this yet, so you may be the ones to help us figure that out. (Update Feb 2016: The RCCX Theory may end up explaining this latter imbalance, due to the CYP21A2 gene mutation lending to hydroxylase 21 being easily overwhelmed. Think “CAH light”.) We often need both magnesium and potassium to properly rehydrate.
9) We often have very young, youthful looking and soft facial skin and makes us look the picture of health! Combine this with some hyper-musculature in some of us, and no wonder you have trouble believing we “feel old inside” as I told my doctor over ten years ago. That same skin may likely need extra sutures or staples or better yet, glue, and extra-long healing time after surgeries, NB. Why this is so, I will never understand! But it is the bane of our existence because it is so misleading.
We never look our age except to the trained eye, who recognizes pain and inflammation, and are often hard-charging, driven, sometimes narcissistic over-achievers. So our complaints once again do not seem to match our appearance or prior history. But it only adds to the misunderstanding and delay in proper diagnosis and treatment for most of us who aren’t born to a diagnosed parent. This is also why it takes 10 years on average to get diagnosed still. It took me over 25. Surely we can do better!
10) The Ehlers-Danlos Syndromes as a whole are not rare, just rarely diagnosed! Yes, most forms are rare, some extremely so. But Castori et al, 2012 stated hEDS may run as high as .5-2% of the population. This has since been revised back down based on the newly tightened 2017 criteria, but even the researchers agree, they don’t really know the true prevalence. Hypermobility itself as a trait runs in 20% or more of the population. I’m willing to bet most are related to someone with a CTD.
Update 2017: Some may now get diagnosed with the new Hypermobility Spectrum Disorders (HSD) since March 2017, and hEDS may be considered technically rare again. But the HSDs are not rare, and generate lots of your “frequent flyers”.
And comorbid MCAS is now being cited to run as high as 14-17% by one leading expert, no kidding! It just looks like/drives many other issues you already treat like anemia, and/or polycythemia, GERD, high BP, refractory high cholesterol, and much more. See the new book “Never Bet Against Occam” by Afrin, L, MD 2016. 
But because this is still so poorly known yet, we may have “Medical PTSD” from so many bad prior visits with disbelieving, poorly informed and sometimes very narcissistic doctors before you as we’ve struggled to be heard and taken seriously and believed. I apologize in advance for any and all patients who “lose it on you” because of this, as that is not acceptable behavior, no. It is not fair or right for us to take out our frustrations on our new doctors, no.
But hopefully the above gives you some insight into where our frustrations are stemming from, and be able to avoid triggering us further. We just need you all to listen a bit more, and be more dialectic, allowing for other possibilities than what all you were told in medical school, which by definition is outdated the minute you graduate alas. My sympathies! Your education is never-ending accordingly, but then, you knew this going in hopefully!
Conclusion
Thank you so much for your consideration of the above and for your understanding and patience! We are complex and fragile sometimes, but manageable with a little understanding on both sides. Thanks for hanging in here with us, and helping us to make it through another day with one of the most painful, debilitating, complex and frustrating systemic disorders I could ever possibly imagine. (I swear, I never imagined I’d be applying my technical pattern-seeking database brain to the medical field).
But as someone once said, truth is stranger than fiction, and I couldn’t begin to make all this up, I promise. Hopefully this will help us all to get on the same “page” of the same “hymnal” sooner. Together, we can handle this lousy condition. Thanks again and good luck!
Jan Groh, February 2015 (Last updated March 2022)
Update July 2018 – Please avail yourselves of the freely available BRAND NEW EDS Toolkit for GPs (PCPs) first published by the Royal College of General Practitioners in London, England in May 2018. (Now hosted by Ehlers-Danlos UK.) I was honored to be tapped to contribute to its development after fellow hEDS patient and NHS GP Dr. Emma Reinhold found and followed me on Twitter a year prior. And share far and wide!
Doctors may also obtain some FREE CME’s about the new 2017 EDS/HSD nosology online here too.
And you might also check out this great article by fellow patient Ellen Lennox Smith on ER Safety Tips for Ehlers-Danlos Patients via Pain News Network 2017.
This is helpful. I have 2 daughters both hypermobile yet dealing with quite different health issues. I am struggling to understand how to get them seen by and under the care of doctors who are knowledgeable about it all and who will provide the help they need.
Thanks for the feedback, glad this post helped. Best of luck to you in your journey – it’s a lot to manage!
This is very well written, Jan. Thank you! I’m thinking I might print it up for my doctor to read as a refresher. I think she forgets sometimes that my complexity can be invisible to most testing methods.
Aw thanks! And yes, feel free! Plus check out my new Resources for Doctors page (where this now lives) if you haven’t yet here:
http://ohtwist.com/about-eds/for-doctors
Cheers!
Thank you very much for all of your information.
I am 46, newly diagnosed, and I have struggled all my life with doctors and people not understanding why I am in pain. Always hearing it doesn’t show anything on x rays or MRIs.
Being unable to take any opioid because of reactions that doctors still don’t believe, ect.
It is comforting to think I am not the only one, and you have given me tips on how to deal with the medical professionals who dismiss my issues. Guiding them and suggesting we look in the direction of those zebras.
There may not be a cure. But understanding and knowing what is wrong still helps.
It is nice to think to myself when I read your information that “It Fits!”
This diagnosis checks all the boxes of MY symptoms that I knew I had and even some that are “Ah ha” symptoms. Those symptoms that you had but didn’t know where connected.
My daughter has it as well, so she can feel like she is now being heard too. (Her diagnosis led to mine).
I have to thank you again. I am so relieved I have found others that understand!
Aw, you’re both very welcome. And I’m so glad to hear that your daughter may benefit by proxy so to speak also. That’s really all I can do – is help re-assure people they’re not crazy, you’re not imagining your pain and experiences, and yes, it’s very hard to get recognized by the medical establishment.
As I like to say, you’re not losing your mind, just your body, smile. And we may not have to fully lose that either before you get some answers. You’re not alone. Speaking of, feel free to join my group either on Facebook here:
Quick EDIT 9/23/25: I have closed my private Facebook group this year, so sorry! Unable to maintain running it for free. Maybe another day!
Or on MeWe, if you prefer a new alternative to Facebook that’s slowly emerging:
https://mewe.com/join/ohtwist
I am 80 years old, who has had numerous surgery for prolapses, 3 miscarriages, and life long bladder & bowel surgery. I was shocked to recently hear that I have Hypermobility. Needless to say I have seen numerous doctors, Consultants over the years, but Hypermobility was never mentioned as to the cause of my medical condition. Why!!!!
I’m so sorry for your long time suffering with no answers, Liz! This is the $64K question, and precisely why I started this blog and tweeting and posting everywhere else I could – to raise awareness for all, but including our doctors! The biggest answer? Doctors are told EDS is really rare, they’ll rarely see it, and once this is bisque-fired into them in medical school, they rarely ever get any newer updated information. Most doctors have never heard of the Hypermobility Spectrum Disorders introduced in 2017 either, so don’t even know enough to diagnose that. And, very few go back to learn more about EDS or HSD or any connective tissue disorders if they don’t have a personal reason to be interested, unfortunately.
WE were just discussing how to get more information into the medical schools (it’s really hard to get the curriculum changed in them, so they just keep repeating old tropes like “EDS is rare”) at the Scientific Symposium Community Day this past Sunday Sept 21st, 2025. Existing doctors already in practice can learn all they’d like through Project EDS ECHO. But just like I never wanted to go back and learn the “New Math” after I graduated in 1985, why would they want to go back and learn the new connective tissue disease info?
Some folks have also cooked up some Massive Open Online Courses (MOOCS) at places like Coursera that offer CME’s, but again, the doctor has to be interested in learning more about it/us. Few will. Medical students can attend Project ECHO even in med school. The more the better. But along with medical misogyny (you’re just a woman/anxious/depressed/it’s just fibro etc.), many of us fall through the cracks for many years (25 in my case from first major complaint at 20), and sometimes our whole lives as in your case and my late parents’. This is every so slowly improving, but no where near as fast as we’d like. But in any case, we need to stop calling it rare – even the rare types. Because we keep putting doctors and everyone else off the scent! Gentle hugs. You’ve really been through the wringer, again I’m so sorry. It’s not right, but I have some hope the younger generations will fare a bit better than us. (I’m 58, and only got diagnosed after a massive onset “cascade” in 2012 landed me in a wheelchair and unable to walk for a year. I still use a cane now 13 years later.) Places like the Norris Lab at MUSC give me hope. Chip Norris pivoted to studying hEDS about five years ago when Dr. Cortney Gensemer arrived for her PhD study. She has hEDS and when he learned that they weren’t really studying it and that no answers were found, he adopted it/us as the lab’s pet cause. They’re producing some amazing results already. She’s now a post-doc and now trains up the next generations of doctors and scientists while doing more bench research herself. Again, you have my deepest sympathy on your needlessly painful journey. I’m so sorry you were gaslit and passed around for so long with no one stopping to ask why besides yourself! – Jan