Heads up change is coming May 2016
 Update February 7, 2017: Via The Ehlers-Danlos Society (aka “The EDS”) on Facebook the full new EDS nosology will be published on March 15, 2017. Meanwhile, some preliminary documents have been shared ahead of time, including one that talks about the new “framework” for recognizing and diagnosing the most common, hypermobile form of EDS.
Update February 7, 2017: Via The Ehlers-Danlos Society (aka “The EDS”) on Facebook the full new EDS nosology will be published on March 15, 2017. Meanwhile, some preliminary documents have been shared ahead of time, including one that talks about the new “framework” for recognizing and diagnosing the most common, hypermobile form of EDS.
So, lots will be changing, and thanks for your patience as I slowly get around to updating everything based on the information as it comes out. And follow me on Twitter and Facebook for the latest always. (See links to the right.) Thanks!
February 18, 2017: The EDS just published an FAQ about the pending new EDS Nosology still coming March 15th, 2017. Patience grasshopper! Patience…
Original post from 2016:
Heads up caveat May 13, 2016: I expect a good portion of the educational information I provide on several of my pages here to change after the results of the discussions of all forms of Ehlers-Danlos Syndrome at the 2016 International EDS Symposium in NYC May 3-6, 2016 finally come out.
I’m waiting on tenterhooks with everyone else at the moment, I’ll update this as soon as I know anything, I promise, I was not able to attend.
Please also note that the former leading US based Ehlers-Danlos National Foundation (formed in the 1980’s I believe) has now partnered with a sister organization (or members of one anyway) in the UK in 2015 to form the new International Ehlers-Danlos Society, or “EDS” in 2016 and the EDNF as we formerly knew it (formerly at http://ednf.org) no longer exists. Yes, some of my links will now be broken accordingly. Please gently apprise me of them at info@ohtwist.com or in the comments here or bear with me as I slowly smoke them out.
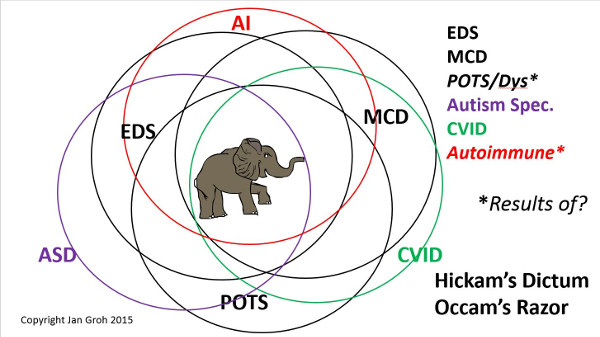
I am particularly keen to hear the Symposium’s take on what constitutes the most common form of Ehlers-Danlos by far, hypermobile type Ehlers-Danlos Syndrome or “hEDS”, formerly called EDS III (3), and often under-diagnosed elsewhere and in the past as Fibromyalgia, HMS, JHS, and BJHS which may all well indeed be variations on the same theme per leading experts since 2009. And equally importantly: how they now suggest diagnosing it.
Editor’s note May 25, 2016: I just got wind that they may have indeed carved out several new SNPs, but these still only account for the minority of patients who exhibit the grossest signs. Here’s reference to a few of them here.
I most dearly hope they are starting to recognize non-bendy patients at the very least somehow and will hopefully ditch or at least rework and rename the overused and highly misleading Beighton 9 pt hypermobility score, since so few pass it yet suffer as badly or worse than those who do. (February 2017: Alas, they chose to ditch the BRIGHTON criteria, with an R and retain the Beighton with an “E” and even increase it one point, sigh, but… at least this will reduce confusion! Stay tuned!)
For newer readers, hypermobile type EDS or “hEDS” is the most common of the soon to be formerly 6 main types we’ve spoken of for almost 20 years now and comprises the vast majority of EDS cases to date , but yet is the most poorly recognized for reasons I go into in depth elsewhere. It is also just the “catch-all” bin for anyone who doesn’t match the currently known more easily diagnosed rare forms, which are much more distinct in presentation (phenotype).
Thus, some people with this label may well have their very own rare form but this just hasn’t yet been discovered. This is likely the case for poor Stryder here.
At this time (May 2016), hEDS can only be diagnosed clinically by elimination of all other known possible forms and related heritable connective tissue disorders (HDCTs) like Marfan Syndrome, Loeys-Dietz or OI etc. (when or if suspected), through careful thorough physical examination and extensive patient and family medical history when available.
This is why I generally recommend seeing a clinical geneticist for diagnosis if possible – they are the most informed on all of these, though technically any doctor can and may diagnose EDS who is willing.
Thus hEDS is a completely subjective diagnosis much like Fibromyalgia which is often diagnosed in its stead (and /or found comorbid depending on whom you ask and what definition you’re using for it), and thus is highly variable in its application from doctor to doctor, even among so-called trained geneticists. Doctors are human too, after all, and quite.
 As most of you know, it is my considered and widely expressed opinion that most doctors have only been told about or remember the grossest signs of the rarest forms of Ehlers-Danlos Syndrome to date, i.e, the low-hanging diagnostic fruit if you will, lending to this being so poorly recognized and rarely diagnosed in their defense. They’re mostly looking for stretchy skin, super bendiness and extreme fragility.
As most of you know, it is my considered and widely expressed opinion that most doctors have only been told about or remember the grossest signs of the rarest forms of Ehlers-Danlos Syndrome to date, i.e, the low-hanging diagnostic fruit if you will, lending to this being so poorly recognized and rarely diagnosed in their defense. They’re mostly looking for stretchy skin, super bendiness and extreme fragility.
But I find many fellow patients just as guilty of the same limited view and thinking, especially if they are very bendy at all – they tend to assume everyone else must be and project themselves onto their fellow patients, and fail to see variations different from themselves and unwittingly invalidate or dismiss many fellow sufferers.
I’ve personally experienced this myself a lot in the last 4 years, and boy are we a stubborn hard headed bunch who aren’t easily willing to change our minds on the whole! ADD and OCD anyone? Literal black & white thinking? Just a touch? Smile.
This is why I’m writing, to help open everyone’s eyes further and suspect it (whatever it turns out to be) much more often.
I’ll finish this lengthy caveat by adding that Dr. Sharon Meglathery’s new RCCX hypothesis proposed February 2016 may well explain why we have not found a single major genetic defect or SNP (single nucleotide polymorphism) that our often overly B&W brains like to latch onto to explain the majority of hypermobile type EDS cases. (Again, not all, sigh. Think grey!)
I don’t count the one very rare AR Tenascin-X haploinsufficiency subset – those folks don’t have to wonder much and are pretty easily diagnosed like the rest of the more rare and easy to spot/bendier forms of EDS. Nor the new SNP found in the Belgian family in 2015 either. These both only account for less than a couple of hundred people in the world total, not many!
But further, the RCCX hypothesis may explain why not all are bendy, and yet we all come with so many additional issues and comorbidities.
Not only might there be multiple SNPs we’re still waiting to characterize on the TNXB gene which may explain the
variability in hypermobile presentation we see once enlightened enough to look for less bendy and stiff patients in addition to the grossly flexible ones, but some may not have any genetic collagen defect to speak of at all.
Instead, they may have issues with their extra cellular matrix (where collagen is formed), and not all to the same degree, if any. Or are related to someone who does in a more obvious fashion. Yet they suffer variations on the chronic constellation of seemingly unrelated allergic, immune, psychiatric and other issues we find so commonly comorbid.
Further, some of us experience changes in our levels of hypermobility over time as I did. Going from extremely bendy like these ladies as a child – I could have been related – to quite stiff in my 40’s when I finally fell apart and got diagnosed. I’m pretty sure I would have been diagnosed with “just” Benign Joint Hypermobility Syndrome in the past.
But I most certainly did not “catch” my hEDS in 2012, it just suddenly severely onset or advanced for as yet unknown reasons. I have my strong suspicions now involving accumulated stress and progesterone based on the RCCX hypothesis. (Many experience this after traumas are severe illnesses like a flu or mono.)
In any case, I trust you are starting to get off the bendy bandwagon with me so I can stop pounding this drum. Again, I’m just asking everyone to be dialectic and allow for more possibilities than perhaps you have either seen or heard of to date – even from so called leading experts. Don’t believe any and everything you read without question – take all with a grain of salt (or more).
And be ready for change, as we continue to learn more and our understanding of the condition evolves as well it should. It should with everything, or we are failing as a species with a supposedly higher intellect IMHO!
Change can be good – it’s just often uncomfortable for many, I know. And I say this just as I’ve started to see the current party line (including not all being bendy) finally slowly become adopted around the world. I expect the upcoming changes (whatever they are) to take a fair amount of time to percolate out accordingly too. I may not even agree with all of them! TBD.
So just please be aware that all of what you are about to read (or have read) elsewhere on my site may be subject to great change in the near future; keep an open mind, and allow for multiple possibilities as we proceed in the future. (Especially my About EDS and Diagnosing EDS pages.)
I look forward to providing clarity as soon as I have some myself. Thanks for your patience and reading!
Jan(droid) 3.0 May 13, 2016
Update February 2017:
Some preliminary articles are leaking out ahead of the full new formal EDS nosology which is now scheduled to be published in the American Journal of Medical Genetics March 15, 2017. A cursory glance shows they are tightening up the criteria for the common hypermobile type “hEDS” mentioned above, and adding a category for those who are bendy and symptomatic, but not quite “clinical” enough (that is, are sub-clinical) called “Hypermobile Spectrum Disorders”, or HSDs, that will likely encompass all of those former old labels (BJHS, JHS and HMS). They also seem to recognize many of us have fibromyalgia as well now. (Finally!)
So… my current info still stands – for now. But I’m expecting to change a good bit shortly after that article comes out. The Ehlers-Danlos Society (the EDS) has promised to make it available for free up on their website as soon as they can. So please be patient, sit on your hands, and let all of us good volunteers and educators have a chance to get it out to you. And… take up any issues you have with it with the EDS or the working groups, not me! (I’m just the messenger, right? Don’t shoot! lol.)
Thank you for your diligence.
Thanks Lynette, I try! Nice to know someone appreciates it…
In principle, if one is older, the physician doing the assessment should take that into account when assessing hypermobility (i.e. enquire if one *was* able to do x/y/z at one point in addition to assessing current levels of hypermobility.
Oh I quite agree. However, my support groups on and offline are full of horror stories of doctors who not only think you have to still be grossly bendy (even when older), but also have extremely stretchy skin. They seem to latch onto the grossest signs of the rarest types only, and ignore the rest for whatever reason. I go into this at length here:
http://ohtwist.com/about-eds/the-brighton-diagnostic-criteria/
Anyway, I’m really glad you’re aware of this, but I find signs of ADD and OCD quite rampant even in the medical community, which lends to leaping to conclusions and minds snapping shut before they’ve finished reading a sentence or letting you finish yours. Very few read the Brighton Diagnostic Criteria fully and thoroughly I find. So I continue to pound the drum…. thanks for joining me.
Hi there,
I am based in Somerset West and have been recently diagnosed by my dermatologist who send me to Stellenobshc university geneticist who confirmed the diagnosis. I am now confused which type of dr to approach to help me with a so-called maintenance plan? I am getting more and more issues.
This is a tough nut to crack for all of us. The key is finding a GP or PCP who is willing to learn as much as they can, and refer you to all the various specialists you will need to address the myriad issues we “enjoy” (sarcasm). Thing is, we “enjoy” so many, that this can be a daunting task for both doctors and patients, so try to “triage” your symptoms and issues, and pick the top 3 for any visit with the GP who will act as sort of the general or “project manager” of your health if you will, hopefully keeping you from ending up with conflicting advice/prescriptions from all the specialists. But ultimately, we end up being responsible for managing it all, and letting any/all doctors we see know any/all medications and treatments we’re taking or engaged in. Yes, exhausting. I would try tapping a local support group (if you can find one )for help finding a good doctor in your region. Try my support page for help with that: http://ohtwist.com/eds-resources/finding-support/
Thanks so much! I got hold of another South African lady – Dotz which I have been chatting to. Just having someone recognise what you saying and who doe snot think you are crazy already make a massive difference.
Wonderful, so glad to hear this – it’s true, sometimes just knowing we’re not alone, nor losing our minds (just our bodies, smile) goes a long way to coping with this enormous syndrome.
Hang in there, glad you’re getting connected up, slowly! Good luck proceeding.