The Hypermobile Red Herring
Or why I think hypermobility is a misleading sign… March 2021
We interrupted our regularly scheduled blogging to bring you a… pandemic. And now we resume…
I had a flash of insight upon watching a video of a talk by Jane Simmonds in 2017 (around the 20:30 mark). I’ve been struggling with the word “spectrum” in Hypermobility Spectrum Disorders since they were invented and described in 2017 – because, a lot of people kept thinking it only referred to the hypermobility or bendiness bit being on a spectrum.
That is, we’ve taken it to be a spectrum ranging from least bendy (and so least visible) to most bendy (most visible). And quite understandably, since a) the very word is in the title itself, and b) all of the literature and criteria to date has been hyperfocused on the Beighton 9 point scale, i.e, your bendiness score. And it’s one of the most visible signs, too. But…
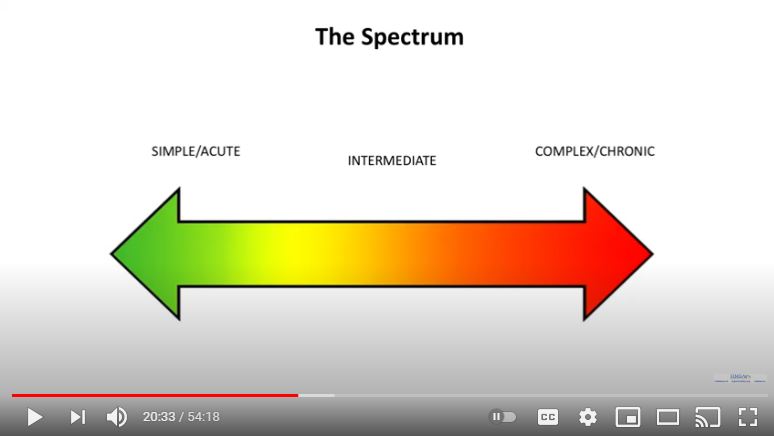
(Screen capture above of a slide from Dr. Simmonds’ talk in 2017 to the HMSA showing a spectrum of ILLNESS, not hypermobility from Simple/Acute on the left to Complex/Chronic on the right.)
…this is not always the case! As her slide above alludes, it can be about the symptomatology – or how “sick” or affected you are.
Which rang a clarion bell for me – this is what I’d been trying and failing to convey in my little Oh TWIST group on Facebook for the last two – nay – THREE years!
SOME people are very bendy (aka hypermobile), but… not very or even at all affected! They would just be called hypermobile, but NOT diagnosed with a Hypermobility Spectrum Disorder. (Emphasis on disorder.) Or a form of EDS. (Same difference IMHO.)
Until and unless they became more affected. That is, symptomatic, with issues that drive them to the doctor. Not just bendiness, which may even just be a fun advantage in their life. (Think Sofi Dossi of AGT, Houdini, The Ross Sisters, etc.)
SOME other people might not be nearly so bendy (aka hypermobile) but… they may be mildly to severely affected with issues with their tissues due to a clearly underlying systemic connective tissue disorder. (Raises hand myself now. I also suspect many late celebrities in hindsight, like Michael Jackson, Prince, Marilyn Monroe and even Elvis. But just my hunches to be clear.)
These people would (or should) be diagnosed with a form of Hypermobility Spectrum Disorder IFF they have ruled out all other possible causes of so many issues with their tissues, including all other forms of EDS, (especially hEDS) and other HCTDs like Marfan, Stickler syndrome, OI, etc. As well as ruled in or out any additional comorbid autoimmune diseases which can greatly mask hypermobility in my opinion.
In other words, you aren’t being diagnosed because you are bendy, in either case! But because you have pain and issues with your tissues in a fairly systemic manner that lends to suspecting a connective tissue disorder.
“If you can’t connect the issues… think connective tissues… “ – anon. (as quoted by Dr. Heidi Collins, MD in her talk of 2014 with this as the title)*
* 2024 note: Dr. Collins has since taken that original video down from YouTube, sigh. But I watched it several times.
So while being super bendy may have seemed to be a diagnostic asset up until now, it’s really just been a big red herring. That is, a misleading sign sending everyone down the wrong diagnostic path by both its presence, AND absence.
Some who are quite bendy but have few to no problems or approved signs technically do not have a connective tissue disorder according to the powers that be. Yet I watch both patients and even many smart doctors fall for someone’s extreme flexibility and hypermobility as a sure sign they “have EDS”. All. The. Time!
But we should really just say “wow, that person is really flexible/bendy/hypermobile!” Period. QED. As we truly know nothing else about them just by looking. Full stop.
And conversely, those of us who, like me, may have either a) stiffened greatly with age and arthritis onset, or b) were born semi-stiff or not very bendy like several I know of, but have loads of issues with our tissues are regularly overlooked and gaslit into thinking its’ “all in our heads”. And we’re “just depressed”~! (My fav, ha.) Precisely because we are not very bendy. (That doggone red herring again!)
No, doc, sorry, but depression didn’t split my SI joint in 2012, nor loosen all my fingers, ribs and toes and cause cranio-cervical instability and settling in me. Nor cause my friend’s severe lymphedema, deafness, CSF leaks, hiatal hernias, ganglion cysts, tendinitis, and chondromalacia patella. (Plus loose or weak teeth, severe myopia and astigmatism and so much more.)
And the even more confusing part? From the annals of stuff I just can’t make up: sometimes, you will have BOTH kinds of people (stiff and or bendy) even in the same family! No, really! Is it anyone wonder our doctors are confused? Heck, even patients are confused!
Do check your impostor syndrome if this is you, especially if you have a larger platform due to minor celebrity. You may mislead a lot of people inadvertently, ahem. Because no, EDS as a whole is NOT rare, just rarely suspected and diagnosed, by everyone. Not just our doctors.
Yes, some forms are quite rare. I happen to be best friends with one of the few with the newly recognized classical-like EDS. (Not the well-known classical EDS aka “cEDS”, but clEDS. There’s a difference.) It took her over seven years to get she and her very affected son properly diagnosed by the best doctors in the world. Finally.
But, you would no more say that cancer is rare because some forms like ovarian cancer are, would you?
No. The collection as a whole is not rare. Even if some forms are.
You special folks may continue to be special and have your own secret handshakes and meetings away from the madding hypermobile crowds. As well you should! You definitely have some unique issues and concerns us “garden variety” hypermobile folks don’t.
But to continue calling the entire collection of forms of EDS with or even without the new categories of HSD “rare” is a serious disservice in my unprofessional opinion. Again, it puts doctors off the scent. And often some patients too. Like a self-fulfilling prophecy.
Heck, Castori et al cited as high as up to 2% of the general population may have #hEDS but a majority are undiagnosed in 2012 here. That’s far from rare. (2% equals 1 in 50 people. Look around you, you undoubtedly know someone with this at that rate, I promise.)
And Tinkle, Castori, Berglund et al cited one study showing as high as 3.4% may have hEDS or a proxy for it in 2017! That’s far from rare! If you then add in the other rare forms of EDS, well, at even just 4% total that’s far from rare.
I’m sorry, but I’m just not that special – and I don’t need to be. Unlike some of my colleagues apparently.
But more importantly: I’m no longer very hypermobile. Even though I used to be, even extremely so like the Ross Sisters shown here. (I have no idea if they had a form of EDS. I mildly suspect so, but again, I cannot say they did. Only that they were definitely quite hypermobile!)
Was I “cured” of my hEDS since I was diagnosed in 2012? I don’t think so! (Trust me, I injure in my sleep every night.) Did I stiffen with age and arthritis so I no longer pass the 2017 criteria? Yes. But just like dear Pluto, I did not otherwise change a spot, nor alter my orbit around the hypermobile sun one iota. I’m still a full planet, dammit! (Smile.) We just changed our minds about what to call me.
Start pulling back, and seeing the bigger connective tissue picture with me. Hypermobility is just a red herring…?
We have you surrounded!


Hi, Jan. Happy Birthday month!
Hyper mobility is such a complex can of worms! It runs in my family. My sisters, 4 of my nieces, my aunt and I all have/had varying degrees of hyper mobility ( and co-morbid conditions, too).
I have always been bendy and “ double-jointed” since childhood…that’s the genetic part. The epigenetic part ( the big “exacerbators”) have been :
1) taking flouroquinolone antibiotics…which can badly damage connective tissue!
2) unawareness that most so-called “healthy foods” are high in oxalates…and oxalates are terrible for people with EDS. Oxalates become deposited in body tissues including connective tissues; also oxalates trigger mast cell storms.
3) Oxalate toxicity used to cause me to spontaneously break out in hives the size of large marbles and some almost ping-pong ball sized hives. Also, frequent ocular migraines, and generalized red itchy skin. What I didn’t know—until about 2+ years ago—is that oxalate was causing me to have these issues.
After learning as much as possible about dietary oxalate and how much it damages tissues throughout the body, I went on a low oxalate diet. Over the past 2+ years, the mast cell attacks have reduced in frequency and severity by about 60%….I am staying on a low oxalate diet hoping to see more improvement.
I also found out that tobacco smoke contains oxalate! Children who grow up in households where parents or guardians smoke end up living in a cloud of 2nd hand smoke full of oxalate. Both of my parents were smokers, when I was growing up. I think this daily, constant inhalation of oxalate in tobacco smoke made my EDS “bendy ness” worse! I also wonder if pregnant women who smoke predispose their unborn children to mast cell & connective tissue problems.
The best website I have found for pointing out how oxalate affects connective tissue ( and other body parts) is Sally K. Norton’s site. ( sallyknorton.com. )
I also hope that you will do lots more research on the virus vaccines prior to getting yours. Take good care of yourself.
Best wishes!
Thank you for this great insightful reply (and the birthday wishes). I didn’t want to touch on why our hypermobility can vary, even within the same family, but… I’m glad you did, and highlighted a couple of big factors.
Both of my late parents chain-smoked my whole childhood, so I was indeed bathed and suffused in the damn stuff until I was 18, ugh. (Thankfully I didn’t take it up myself!) Anyway, while my hypermobility spectrum disorder and hEDS signs runs clearly through my family and on both sides, I do think these epigenetic factors may well play a role in my relative ‘severity” of problems when I fell apart so badly at 45 in 2012. (Whence this blog.)
I also think we have a lot of transmitted inter-generational trauma of other kinds too lending to our trouble also. But that’s a really big can of worms for another day. In other words, while a small minority do have some clear genetic SNPs driving their hypermobility, those of us with the most common form: hEDS and/or the new category of HSD (if you’re not bendy AND/OR troubled enough) may well enjoy the effects of things like oxalates etc. on our systems.
I think I also react to oxalates, though a bit more mildly than yourself. I say this as I do add 1 single kale leaf to my celery juice every other day with no great ill effects. (Or maybe not that I can detect yet, smile.) But it is good information to know.
(ETA: I did have bigger reactions and passed out a lot more when I was juicing more kale more regularly a few years ago. I finally connected the dots and stopped emphasizing the kale, smile. Whence I just use one leaf now, and only every other day tops. Again, I think my reaction is milder than yours, touch wood quick.)
I definitely think my parents chain-smoking played a role in my current health among many things, including my unhealed childhood complex trauma. (I was bullied from kindergarten until I left highschool at 17, and neglected and verbally abused by both of my parents before age 10.) So many factors… so little time. My older sister seems to be doing better than I. But she was born 4.5 years before me so… who knows if my parents coping skills weren’t much better until I came along. (They were 40 years old when I hatched.)
I see signs of both autism and ADHD AND CPTSD in my late mom in 20/20 hindsight, and I think she could not cope with the sensory issues and pain she experienced, and lost her shit on us because of this. (She was orphaned by age 8 herself and shoved around what passed for foster care back in the 30’s, poor thing.) So… who knows? But I do think we are a complex package of nutritional deficiencies, sensitivities and a SNP or two (or three) still TBD. (I think if it was an easy single SNP we’d have found it by now which is all TEDS cares about. I’m pretty sure it’s more complex for the reasons we’re both describing as well as a la the RCCX Hypothesis here: https://rccxandillness.com .)
Anyway, thanks for sharing your additional wisdom and insight on the oxalates. I’ll drill down when I can thanks.
Wow, Jan! I see some interesting parallels between your familial/generational factors and mine.
My mom was the abused child of an abused child. For my mom, it was so bad that she grew up to have agoraphobia, some type of depressive narcolepsy and a personality disorder. My dad was on the autism spectrum; Aspergers but very intelligent and high functioning. I am pretty sure I’m also an aspie. I think my older sis is also an aspie. My younger sis seems to be the most normal one in the family.
My parents were unable to love us. They did the best they knew how at the time.I will leave it at that.
The RCCX theory is fascinating and brilliant! I wish more people knew about it. I really think that both my parents carried this “package” of gene mutations. I think “mutants” have a way of ending up together and having unique, gifted “mutant” kids.
Jeanne H
I completely agree about us being highly attracted to each other for whatever reason(s). I’ve yet to find either a) any fibro patients who don’t also have signs of an actual CTD, usually involving hypermobility to varying degrees, for one thing and b) most who do get diagnosed (dxed) with hEDS/EDS etc find they have signs on BOTH sides of their families, once you drill down far and hard enough. (Read: get past all the denial, usually on the male side.)
I wrote about all the commonalities I was seeing here, in case you haven’t seen this article yet. It was my writing and insights that led Dr. Meglathery to drill down in the literature and come up with the RCCX hypothesis to possibly explain it for some of us:
https://ohtwist.com/the-chronic-constellation
Cheers.
This is brilliant! Thank you so much. It really resonates here as I have one child that barely meets the criteria but is terribly impacted. Getting doctors to take her seriously has been an uphill battle. Then I have another child who not only scores an eight, if they added shoulders, ankles and other movements, she would score more. Despite being far more bendy than her sister, she’s absolutely fine.
TY! I rest my hypermobile case! lol. I so hope doctors catch on and stop thinking in rigid black and white terms. (All bendy, or nothing.)
Thank you for this article and thank you Jeanne for bringing up oxalates. I discovered I cannot eat Kale or raw spinach when I developed a kidney stone after a bad flu several years ago. (Dehydration + oxalates) I ate them occasionally since then but decided why punish myself when I didn’t really like it? (Feeling rebellious and then guilty about it is a lifelong thing for me).
I also grew up with a smoker and once away from it as an adult discovered that I felt nauseated when I was around it.
Also with the generational pattern of abuse which I tried to break. Every step away from it is an improvement.
I really appreciate the links and will explore them now.
Happy belated birthday Jan!
You’re welcome. And with that history, you might also look into MTHFR SNPs (genetic defects) in your 23andMe or similar data. Many with one or more of those find they are sensitive to cigarette smoke. (That’s another whole deep rabbit hole of its own. I highly recommend following Dr. Ben Lynch for more on this: https://mthfr.net ).
And good sleuthing on the kale! More proof one diet type really doesn’t work for all. Thanks for the birthday wishes!
It’s interesting to find somebody else who has fallen afoul of the change in diagnostic criteria.
I developed back pain at the start of 1997, it hit suddenly and literally swept my legs out from under me. I’ve had brief spells since then when my spine and hips were strong enough to support my weight, and when the pain of standing didn’t overwhelm me, making me black out, within a few moments. I rode the “psychosomatic” roundabout for six years (relatively short I know) when a friend asked if doctors had considered “Hypermobility Syndrome”. They hadn’t, I asked and I was sent to a consultant who thoroughly examined me (Beighton 9/9 although my knees were borderline) and told me that yes, I had “Hyper-Mobility Syndrome; Ehlers-Danlos”. She explained that the second term was just a fancier name, I would only need to remember HMS.
Fast forward 15 years, which were unfortunately filled with mistreatment, my husband would refuse to get me help when a dislocation wouldn’t go in easily, he prevented me getting to appointments with doctors and specialists and, over time, he made it increasingly impossible to leave my bed.
Eventually I found the courage to leave, which was when I found out that the consultant had written Hypermobility Syndrome but now that doesn’t mean EDS. So I needed to see somebody to be rediagnosed. Just one problem. My wrists dislocate frequently and it seems they have some permanent damage, so they aren’t as flexible. My thumbs (which used to bend to touch my wrist as the test requires, and the other way round too) dislocate at the slightest push so I couldn’t bend them around without causing serious damage and I have Duputreyn’s contractures on a number of my fingers (btw, that’s another thing which is often associated with EDS for your list). I also have contractures in my legs, as a result of being confined to the bed. You can imagine what all this did to my Beighton score (and it didn’t help that she tried to do the hands on the floor bit WHILE I WAS WEARING A CORSET!) So even though I had all the other symptoms (except the bruising, I have very deep veins and for some reason, I just DON’T bruise, I’m an albino zebra) she said my Beighton score showed no sign of hypermobility (except my elbows) so it couldn’t be EDS. And, by the way, what moisturiser do I use? (Yes, she decided my velvety skin must be due to good toiletries!) When asked what she thought WAS wrong with me, she said that, if I MUST have a diagnosis, she supposes it’s fibromyalgia.
Since I “stopped having EDS”, I’ve discovered more of my current health problems and/or things which I’ve had all my life which are actually commonalities, or are tied to, EDS. I stopped counting after I had twenty-five or so of the issues you listed in each of your “Unofficial Comorbidities” and “Signs” lists. (I don’t have the Gorlin’s sign (I can’t touch my nose with my tongue) but I do have a severely reduced, almost non-existant lingual frenula (that flap of skin which tethers the tongue to the base of the mouth) and lacking that and/or the labial frenula, which attaches the lower lip to the gum, are signs I thought you might like to add to your list. Apparently those who know about these things considered including this as part of the minor criteria in the 2017 diagnostic review.) I even learned a new one today, “EMF sensitivity – and tendency to “blow” nearby electronics”. I’m so glad I’m not the only person that happens to. It drives me mad how quickly electronics wear out when I’m near. It gets kind of pricey, lol!
So now I feel rather lost. I’ve seen physiotherapists since my “reassignment” (to try and help regain strength in order to prevent all the dislocations, I’m sure we’ve all been there) and they are all left speechless that somebody could think I’m NOT hypermobile. They aren’t doctors so there’s nothing they can do about it (and come to think of it, how can a junior doctor overrule a consultant? Surely if I was diagnosed as hypermobile back then, my Beighton must have been 5 or more and if things like the velvet skin and factors which don’t change would, with a higher score, be classed as hEDS now, they must have been the same then. I’ve asked for a second opinion but now I feel like I’m just getting treated like a hypochondriac. I’m very interested to see what will come out of the review this year but it doesn’t look like they will be looking at the problem of people who DO have EDS but lack the hypermobility. Oh well, fingers crossed.
I’m going to look into oxalate toxicity, I’d never heard of that before.
Arrrgh, I’m so sorry! THIS IS WHY I WRITE ABOUT THIS STUFF! Doctors have such a limited narrow view of EDS, they can’t see the forest for the damn trees! Sorry to swear, but it’s beyond aggravating at this point and bordering on malpractice. Seriously, if someone told me they use to be a 9 on the damn Beighton (with an “e”) 9 pt bendy scale with enough issues to be diagnosed with “HMS” back in they day, and then described what you have about the joints being too unstable to bend now, or as in my case stiff (and breaky – if you did push me, mine would come apart like yours now, so I don’t ), I think I could put it all together and say “gee, walks like a duck, quacks like a duck” and… as I like add: “suffers like a duck”. And I would probably conclude it’s a duck! (EDS). SMH. Whence this post, which I’m glad you found. And yes, I was beginning to suspect Depuytren’s Contractures might go with this, so thanks for the added push. I’ll add that to the long (and slowly growing) comorbidities list.
I know it’s a big ask, but… if you can take a rest, gather your strength and regird your loins, I would try again for a proper diagnosis if you can. It’s just such a very fickle system: some people are diagnosed bang, easy, by a rare smart doctor, or on a good day, etc. The rest of us? Chopped liver, stuck swirling around the drain of suffering in perpetual diagnostic limbo and dismissed and denied proper care. Grrr.
Glad you found my site and some minor validation at least. I can’t diagnose you but DO strongly suspect you of an EDS, yes, most likey hEDS but I can’t say without a proper full history of you and your family. And yes, it’s odd that you don’t bruise easily but… that’s more to do with capillary strength than the depth of your veins. Anyway, welcome to the “family”. I consider you a member.
Hey there! I see your in Oregon, would u mind sharing me a good dr to see around here? I’m 35 and basically just diagnosed myself a few weeks ago. Lol. If anyone knows my body I do! And I know something is def wrong! My whole life has been a mystery as to why my body is starting to fail me at this young of age. The list goes on of my symptoms. I’m getting my blood results back this week but I’d love to connect with a dr who knows about this. Thanks so much!
Hey Kristina, thanks for asking. Ironically, I haven’t kept in the local loop on good doctors for a while – I’ve had personal matters to attend to (including this, my blog), but… I would encourage you to inquire of the Oregon EDS group here:
https://oreds.org/index.html
We have long lacked for many if any good doctors who can (or more importantly *will*) diagnose besides Dr. Schirripa in Bend, Oregon – a medical geneticist in private practice. He’s wonderful, but both spendy (depending on your insurance), and alas, he’s really poor on follow up / follow through with reports, and referrals after you have your great initial meeting. (He’s super warm and fuzzy, and loves to geek out and will recognize ALL the comorbidities, unlike most, in person. It’s just after that – out of sight, out of mind alas.) So… that’s all I’ve got for now. I hope you can get more from OREDS. Dr. Schirripa is here fwiw:
https://cocgc.org/
Tell him I said hi!
I have dup contracture as well. I had surgery to my right hand which ended up giving me lymphedema of the right area and hand chest wall. ( prior breast cancer lymph node removal ) all the surgeries caused RSD of my right arm.
I was sorta flabbergasted when I didn’t pass a simple Bright score. Did the physician not notice that my pinkie on the right hand was permanently fused bent by scar tissue and no way I can bend it backwards . passed on both thumb to wrist and if they asked me I could have done and passed the Marfans score.
Then I end up with familial dysautonomia ( Riley – Day) and CCI, raynauds. My birth mother has MVP, numerous strokes. Passed the EDS 3 diagnosis as I did in the past, but now I’m concerned about the vascular issues since my grandmother on my maternal side died at 42 waiting for a heart valve replacement . She had been a pianist and was in a wheelchair due to osteoporosis of the spine.
I have a MTHER variant and am missing both CYP2D6 allele.
I wish I could afford the testing for vascular and classical. I’m 65and it’s so frustrating not being able to find a single doctor that takes Medicare.
I meet these new doctors who are treating people w EDS and mainly have learned from their patients. Most are younger and are being treated pre damage. I’m at the needling a power wheelchair .
Hi Claire, you are exactly the type of person I wrote this post for! It is sad and rather frustrating how doctors can’t think outside of the hypermobile Beighton Score box, sigh. I’m so sorry. I just added Dupuytren’s contracture to my comorbidities list not long ago actually. So sorry you have this! (It sounds quite painful.)
Interesting that you ARE diagnosed with the rare Riley-Day form of familial dysautonomia – I haven’t seen that outside of people with soem of the rarest forms of EDS. So… hmmm. You may have a leg up with a geneticist if you could gather enough family history to convince them to test for one of those forms. One of my friends turned out to have the newly recognized “clEDS”, aka “Classical-like EDS”, NOT the same as Classical EDS (aka “cEDS”) which is not that rare at 1 in 5000. clEDS is more like only 500 people in the whole world are identified yet. (Very rare, or at least rarely diagnosed yet.) She had to go to several of the top specialists in the world (US) to figure that all out too. I’ll note she has extremely fragile skin and soft inner tissues – her esophagus tore in her 20’s. And she and her son don’t feel pain very easily, so injure more than they should from this. Has that been the case for you?
MTHFR runs in almost 50% of the world’s population, so that’s not that uncommon – but I definitely think it doesn’t help, either. I don’t quite agree with Debby McQueen who thinks it causes hEDS. If that were true, then 50% of us would have hEDS and even I (who think it’s around 10% or more of the population, unscientifically) don’t think it’s THAT common. But again, it definitely lends to our experience fo overall chronic illness alas. Glad you figured that out. Debby’s blog is here for what it’s worth:
https://mthfrheds.com
I note that she misleadingly lumps all MCAD under the umbrella term “Masto” which is incorrect. MCAD is the umbrella term under which all forms of masto fall, along with the newly recognized (since 2016) MCAS that everyone struggles so hard to get diagnosed also.
I feel you on the Medicare bit. We sort of get the least pick of doctors and they are rarely up on these things alas. And, for what it’s worth, I also needed apower chair nine years ago – at 45. I’m happy to say that high dose vitamin C*, magnesium and warm water therapy got me back out and walking more again. But I still use a cane at 54. (My right leg is permanently loose in the socket. Funny, my late Aunty’s was too, heh. We were definitely related.) My point being: there’s no shame in needing a power wheelchair. But… I hear your frustration. You’d rather not need one.
* I have to admit to being lucky to both a) tolerate cheap corn based ascorbic acid in OTC Vitamin C unlike many and b) have half decent digestion, also not common in EDS (all flavors). So I’m lucky on this front. If you are allergic or sensitive to corn, try either Sago palm based C, or moringa leaf powder in your food. (We often react to citrus from comorbid MCAS which is high histamine.) And one friend with hEDS went from bed bound to running again with Vitamin C injections she describes at length here:
https://lessflexible.com
I’m working up to this – I’m too poor to afford them at the moment.
Regardless, I dearly hope to prise open more medical minds to think outside of the Beighton score, sigh. Hang in there!