The Privilege of Diagnosis
Perspective: for background, know that I’m a well educated, hyperverbal and hyperlexic white allocishet normative woman from the western US, with all the privilege that entails, despite my poverty. I just want to recognize that at the start of this post about my diagnostic journey. So many others have even fewer resources to work with, including even the internet in some cases.
It is the 10 year anniversary of my formal diagnosis of hypermobile type Ehlers-Danlos Syndrome (aka hEDS now, formerly EDS III), Valentine’s Day, 2012 at almost 45 years old by a medical geneticist. Yes, we ruled out a couple of the rare types that we suspected too, based on my family history. (Blood tests came back negative for vEDS and kEDS.)
This, only after I suddenly went from walking to wheelchair in barely three weeks that January, blowing all of our hairs back and finally getting my doctor’s attention! Yes, it was as shocking and as painful as it sounds. I’m SO glad I didn’t know what was coming the previous year or I might have opted off the ride! All after my most successful year of database programming ever, sigh.
No, I wasn’t “just depressed” as they’d pegged me up until then. I did have dysthymia, but that didn’t explain any of my pain. Yes, I had a very real, familial connective tissue disorder that had plagued me from birth, but had been dismissed until I was 45.
All because we keep saying it’s “rare” so doctors don’t think to consider it, and dismissing AFAB people who express any pain or fatigue as “just depressed” or “anxious” (uhm, we may be those things too, but you keep dismissing all of our very real physical issues!) Medical misogyny and psychologizing is alive and well, alas.
Near misses through the years…
The closest I came to a physical label previously was a diagnosis of CFIDS back in 1993, which didn’t quite fit fully either. Oh, I had bouts of fatigue, and some low-grade unexplained fevers, often leaving me in bed, but… they never lasted long, quite unlike the people I met in the one and only support group meeting I attended for that.
Many were in wheelchairs and some hadn’t left their houses in months! I might go down for a couple days tops, but I was nowhere near as disabled as they were. So I got on with life as best I could despite having all the signs of fibromyalgia too, whatever that truly is. (Ask ten people, get ten different answers which is why I’ll let you pick your favorite answers from Google yourself.) I was one tender-point shy of the cut-off for that diagnosis then too, sigh.
Story of my life, never quite fitting in or belonging anywhere… but I digress.
Fast forward to 2007 when I injured my upper back twisting while collating paper medical records at a busy urban hospital after only three weeks at it, and my massage therapist kindly suggested I might be “hypermobile”. (I took out T3-5 which are loose and quite painful to this day.) This really bummed me out because I truly loved that job, too!
I had never heard of this term, but a quick Google netted me the Hypermobility Syndrome Association of the UK, and an article by rheumatologist and Hypermobility Syndrome and EDS specialist Dr. Rodney Grahame at the time that described me to a “T” and I knew I’d found my people!
Alas, my doctor dismissed my suspicions once again with a wave of his hand saying “you probably don’t have that!”, until my body finally hit us both over the head with a virtual brick in January 2012 and sat me up (or down, I guess) in my wheelchair and made it patently obvious I did too have that! Or some variation of it, as I slowly fell down the rabbit hole.
No thanks to my family who were all either in denial, or dead. (My alcoholic parents passed relatively young in my life without being diagnosed, either of them, despite both having plenty of signs retrospectively.)
Someone tipped me off to Facebook groups in December 2011 as I was starting to weaken thankfully, and the rest is “herstory”. I hate Facebook believe it or not, but the groups have literally saved my life, repeatedly. (Thank you all!) So much so that I’ve even started my own Oh TWIST group. (Come on in the water’s fine! Diagnosis not required, since they’re so rare. I’m also here, on MeWe if you prefer to avoid the former platform.)
I found “my people”, and was able to arm myself with sufficient medical info and literature to make my case, and got my diagnosis from the geneticist on February 14, 2012. (Yes, a memorable “date”, ha ha.)
Happy Tenth “Diagnosiversary” to me!

Whence my domain name. (Oh, That’s Why I’m So Tired – Oh “TWIST”!) As in, no, I may not have CFIDS, aka Myalgic Encephalomyelitis / CFS now thankfully, but I have since uncovered the myriad causes of all my pain and fatigue – finally. (My heart goes out to those who actually have ME/CFS, much less both EDS and ME/CFS like Dr. Ron Davis and his son – no kidding.)
My story is not that unique, unfortunately. And I’m even luckier than many still twisting in the breeze and being kicked around the western allopathic medical system or abandoned, dismissed and gaslit by their families and or doctors.
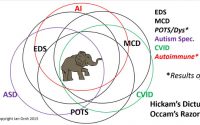
Between everyone calling EDS “rare” – when combined with HSD (or its proxies)it may run as high as 3.4% of the general population, making it far from rare, and doctors only remembering outdated descriptions of the grossest signs of the rarest types, and the aforementioned medical misogyny and psychologizing, well, very few manage to hit the medical radar and get diagnosed even still.
Non-binary and trans people are also missed for any or all of the above reasons too. (Apologies if I’ve left anyone out, please kindly apprise me in the comments thanks.)
And, we tend to come down with any number of either secondary and co-occurring conditions (arthritis, IBS, fibromyalgia, one or often more autoimmune conditions and more as I illustrated here), and all of these end up masking our underlying hypermobility as well.
Denial is not a river in Egypt…
That’s if doctors are even willing to see past our very real, understandable and often also secondary depression and anxiety too. They truly seem happy to stop with a psychological diagnosis, and write everything else off to being “just depressed”. Ouch. (My diagnostic journey above is a typical result.)
Don’t get me wrong – we do need to treat our anxiety and depression! But we need to stop dismissing any and all other complaints in the process! (Aka “psychologizing” physical complaints.) Talk about diagnostic overshadowing, whew…
And our families don’t always help either. Mine were in denial of any major problems, despite gross and obvious signs and suffering on both sides in 20/20 hindsight. My late aunt Sr. Kathleen Groh (a nun) was diagnosed by her nursing home doctor sometime in the last ten years (without telling anyone) before she died. (I just happened to ask about it during a visit in 2013 and learned then.) And I have a cousin on my mom’s side now diagnosed by US EDS specialist Dr. Brad Tinkle with whom I connected about five years ago. (Not in time to help my diagnostic journey in 2012 though.)
Everyone else was either too stoic, or also dismissive of my issues and me too. So I thought I was just a wimp and a weakling who needed to “Jan up” if you will all those years. I know my employers and doctors sure all made me think so. Most friends too. We are such an ableist society!
I also happened (just happened) to pick all the right hobbies for helping to support and mask a connective tissue disorder all those years – synchronized swimming from ages 9-13, cross-country running in highschool, biking and folk dance after that, and flute playing from age 10 on. All incredible therapies in their own rights that probably helped hold me together and help me mask and pass so well until 2012. (And continue to help me as I recover, ten years on.)
Of course I couldn’t have EDS – I was too active! And I had inadvertently picked activities that helped strengthen my core muscles among other things, helping me to hold together much better and longer than I might have otherwise.
That is, again, to mask and pass – and not pass out!
So I actually feel lucky. Truly. As painful as 2012 was, and life has been ever since, I at least got taken seriously – finally, for once in my life. It should not take 25 years of related symptoms and physical complaints and landing in a wheelchair to finally get diagnosed! For anyone.
And that’s just my EDS diagnostic journey.
My parallel journey…
I was just finally diagnosed as autistic with ADHD (aka “AuDHD” per Twitter) just last summer, 2021 too, at 54 years of age. That’s a whole other journey of its own to share another day, oh my. But the parallels are eerily similar: getting dismissed as a woman, psychologized, mis- and under-diagnosed along the way. And made to feel like an imposter. (And still getting bullied to this day, sigh.) Yet still privileged, as a white woman.
Which is also not at all rare, and a sadly all too common experience for many others too. Once again, doctors and other practitioners (therapists, counselors, teachers and more) tend to only recognize the grossest most obvious or most stereotypical signs of a subset of autistic people based on outdated literature, so… you shouldn’t be surprised.
The barriers of racism for too many…
I’ll just add that if I, a white woman in the western US had this much trouble getting diagnosed with EDS, God help everyone else in less privileged shoes. In the US, Black people’s pain is regularly dismissed, especially in women. Black people in the US may tend to avoid seeing doctors based on our past horrible history of experimentation without consent. And cultural biases and systemic racism within the western medical system can all lend to dismissal of very real issues as described, too.
And all other non-white people in the US share variations on the above challenges – discrimination, underfunded health care, lack of access and support.
And most of the latest and best medical literature is all in English and a few other European languages (French, German, Italian, Spanish and maybe some Russian to my knowledge), so I have no idea what other parts of the world have to work with, aside from Dr. Jaime Bravo’s great site and care down in Chile. Is anyone translating anything into Mandarin, Japanese, Thai, Pashtun, Urdu, Armenian, Swahili, Indonesian, Turkish or Hindi, to name just a few? I dearly hope so. (Admitting my ignorance here. I did find one site in Hebrew, thankfully via TEDS.)
I just know we have a long way to go before the average time to diagnosis drops below 10 years, even still. I will take my blessed privilege, and run, figuratively speaking. (More like hobble!) And continue to keep my site as up to date as possible for all of our sakes to that end.
To your health,
Jandroid 3.14159265


Hi Jan and Happy Diagnosaversary! I really enjoyed this post ( I enjoy all of them). Thank you for the great links and information. I have a lot of reading to do…
This time, I learned quite a bit more about dysautonomia. I have had it since childhood through the present. The kicker is…my mom had it too. I recall her needing long naps every afternoon of her life. She slept 8 or 9 hours at night and maybe 3 or 4 every afternoon. Our lives, as kids, were ALL about being QUIET so mom could sleep. She was the queen of tired. I only just learned that this is part of dysautonomia.
The long journey of navigating through medical misdiagnosis, medical ignorance, familial indifference, is mind-blowing. (Sigh…) I’ve had my share of that since 2008. I ended up needing to do my own research…probably hundreds of hours of it…to figure out what was going on with my body…and remedies, if any. It’s a work in progress.
Thank you so much Jeanne, I’m so glad my writing both resonates with you, and helps you at all on your diagnostic journey(s). And oh boy do I relate to napping parents. My dad did work full time as an engineer, but… he always took an hour nap after getting home from work, and we had to tiptoe around and not bother him until he was ready to get up. (Sometimes not even then.) And my poor mom, too, I now know why she had so many issues, and drank so much for her pain. Neither of my parents got properly recognized or diagnosed with anything besides depression, anxiety, arthritis and alcoholism. 🙁
(Nothing wrong with napping when you need! Or taking a sensory break eitehr. Just, be clear that it’s not the kids’ fault, and that you will be happy to attend to them/play/look at school work later when you can. Mine never did this, we were mostly latchkey kids with parents who went through the motions, alas. So I just felt unloved.)
I think most of us still end up doing our own research. The number of people recognized by doctors is still a very very small percentage, which is why I write so much – to hopefully help everyone (patients, families, and doctors) alike recognize this whole package sooner. And empower you all to diagnose the relevant bits much sooner than we are, yes. I wish you the best of luck on your journey. You are so not alone!
PS Good news: The EDS Toolkit for Doctors still lives – just over at the EDS UK site now, here: https://gptoolkit.ehlers-danlos.org