Diagnosing Ehlers-Danlos Syndrome

Boy is this a hot topic! Naturally, since it’s so hard to even get a doctor or doctors to listen to us and recognize our vast collections of symptoms (see last) as a possible systemic condition, we naturally want an easy test to take and be done with this painful journey and have an answer. (Our often Aspie brains want this too, natch.) Well, I’m sorry to break it to you, but there is no single test for all types of Ehlers-Danlos Syndrome yet. Not only is there not just one single type of EDS to begin with, but there is no single tissue marker for the most common type of EDS, known as Hypermobile Type EDS or HEDS yet. So even if you got Whole Exome Sequencing (WES) testing or some other thorough panel or set of tests (like the TAAD panel at UW), you still cannot rule out (or in) all EDS this way. HEDS is only diagnosed clinically once a knowledgeable medical doctor (preferably a geneticist) rules out all the more serious forms of EDS, and all other Heritable Disorders of Connective Tissue. You are then left to diagnose Hypermobile EDS (or HEDS) clinically via thorough physical exam and extensive family history where available. (A bit hard for adoptees, sorry).
This latter is done using the Brighton Diagnostic Criteria (not the Beighton 9 pt scale only!) presently for lack of a better tool at this time. The Brighton Diagnostic Criteria INCLUDES the Beighton 9 pt Hypermobility Scale. But you do NOT have to score high on the 9 pt Hypermobility Scale to have EDS!! In fact, you can score a ZERO on the 9pt Beighton Hypermobilty Scale and still have EDS! (It’s just an epidemiological field test for the trait of hypermobility developed over20 years ago by Professor Peter Beighton). Hypermobility itself is just a trait, like red hair color, and in itself is generally benign. EDS is hypermobility plus symptoms, or issues that ultimately drive you to see the doctor a lot (for those who aren’t in denial). This is the whole reason the BRIGHTON (as in Brighton, England) Diagnostic Criteria were developed – the leading specialists realized they were missing a lot of older and non-bendy (non-hypermobile) patients who are either no longer flexible, or never were, but who all have common extra-articular (non-joint-related) issues and signs of a collagen defect. (Stretch marks, odd scarring, thin or stretchy skin, myopia, varicose veins, tendonitis, hernias, prolapses, etc.) But they did not have obvious signs of one of the more rare types of EDS leading to testing for those.
For those who missed it in my About EDS post, there are six main types of EDS presently recognized, but new mutations arise all the time. (Life’s messy, there aren’t enough paper towels to clean it up I’m afraid, and never will be, sorry to all who jones for tidiness and order). These are now known by their descriptive names to avoid confusion between the collagen number type and the old former EDS number type. Listed here in order of rarity, from most common to most rare:
- Hypermobile EDS (HEDS, formerly Type III) – can only be diagnosed clinically yet, sorry, except for the rare Tenascin X subtype
- Classical EDS (CEDS, formerly Type II) – does have a tissue test, but often thought unnecessary since like HEDS, it’s usually not lethal
- Vascular EDS (VEDS, formerly Type IV) – has a definitive blood test now via UW Collagen Diagnostic Lab (see below)
- Kyphoscolioitic (KEDS, formerly Type VI) – pretty rare, characterized by more severe kyphoscoliosis and keratoconus among other things
- Arhtrochalasic EDS (AEDS, formerly Types VIIA and B) – quite rare, in only a few families in the world
- Dermatosparaxis type EDS (DEDS, formerly Type VIIC) – very rare, characterized by extremely stretchy skin that then doesn’t tend to “spring back”, and remains in folds
Most cases (HEDS and CEDS) are autosomal dominant, meaning that if one parent carries the defective gene, you have a 50% chance of passing it on to a child. But some are recessive, so are even more rare, thankfully. (Both parents then have to carry the recessive gene for the child to express the defect also, so it’s closer to a 25% chance). There are single tissue markers and tests for all but the most common type, Hypermobile EDS now. As already noted, this latter can only be diagnosed clinically by ruling out all other types of EDS if suspected, as well as other HDCTs if suspected, and then studying the patient’s family and physical history thoroughly. You can (and should!) learn more about the main types of EDS here.
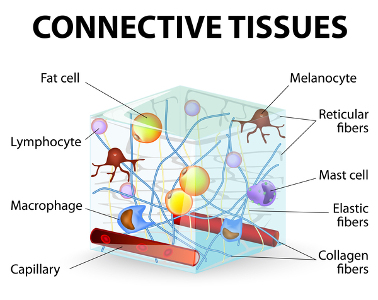 Further (again) there is a LOT of crossover in symptoms between EDS and some of the other Heritable Disorders of Connective Tissue like Marfans Syndrome, Osteogenisis Imperfecta (OI, aka “brittle bone disease”) and Stickler’s Syndrome. (Why you should seek out a knowledgeable medical geneticist for diagnosis where possible). And, there is a LOT of crossover of symptoms and signs between the various TYPES of Ehlers-Danlos Syndrome itself. (They all involve different collagen defects in different types of collagen, and not all are identified yet as already noted). So it’s not an easy, cut-and-dried thing to diagnose at all. Almost everyone with EDS has fairly visible blue veins, easy bruising and more. And almost everyone wonders at some point if they don’t have Vascular EDS (VEDS, formerly Type IV), a dangerous rare form that lends to more shortened life spans than most from arterial or inner organ rupture. I’m happy to report very few of us will/do, thankfully. And I’m happy to report that VEDS patients are defying the odds and living even longer on average than ever before, thanks to improved modern medical care. But by all means get the blood or tissue test done to firmly rule it in or out if you do suspect it based on clear personal or family medical history. But most don’t need to worry about this unless you have a distinct family history of early demise from aneurysms or internal organ or vascular rupture as noted here at the UW Collagen Diagnostics Lab:
Further (again) there is a LOT of crossover in symptoms between EDS and some of the other Heritable Disorders of Connective Tissue like Marfans Syndrome, Osteogenisis Imperfecta (OI, aka “brittle bone disease”) and Stickler’s Syndrome. (Why you should seek out a knowledgeable medical geneticist for diagnosis where possible). And, there is a LOT of crossover of symptoms and signs between the various TYPES of Ehlers-Danlos Syndrome itself. (They all involve different collagen defects in different types of collagen, and not all are identified yet as already noted). So it’s not an easy, cut-and-dried thing to diagnose at all. Almost everyone with EDS has fairly visible blue veins, easy bruising and more. And almost everyone wonders at some point if they don’t have Vascular EDS (VEDS, formerly Type IV), a dangerous rare form that lends to more shortened life spans than most from arterial or inner organ rupture. I’m happy to report very few of us will/do, thankfully. And I’m happy to report that VEDS patients are defying the odds and living even longer on average than ever before, thanks to improved modern medical care. But by all means get the blood or tissue test done to firmly rule it in or out if you do suspect it based on clear personal or family medical history. But most don’t need to worry about this unless you have a distinct family history of early demise from aneurysms or internal organ or vascular rupture as noted here at the UW Collagen Diagnostics Lab:
“1. Possible diagnosis of EDS type IV: The vast majority of probands in families with this form of EDS are identified on the basis of a major complication either bowel perforation or vascular aneurysm or rupture. The International Ehlers-Danlos Foundation Advisory Board set the following guidelines for determination of the clinical diagnosis of EDS type IV. DNA-based testing is recommended for those who meet these guidelines. Note, however, that individuals with nonsense mutations of COL3A1 are less likely to have similar physical characteristics. The clinical diagnosis of EDS type IV is highly suspected when two major diagnostic criteria are present:
Major clinical diagnostic criteria:
- Intestinal rupture
- Arterial rupture
- Uterine rupture during pregnancy
- Family history of the vascular type of EDS
Minor diagnostic criteria alone are not sufficient to warrant the diagnosis unless identified in an individual with a major criteria.
- Thin, translucent skin (especially noticeable on the chest/abdomen)
- Easy bruising (spontaneous or with minimal trauma)
- Characteristic facial appearance (thin lips and philtrum, small chin, thin nose, large eyes)
- Acrogeria (an aged appearance to the extremities, particularly the hands)
- Hypermobility of small joints
- Tendon/muscle rupture
- Early-onset varicose veins
- Arteriovenous carotid-cavernous sinus fistula
- Pneumothorax/pneumohemothorax
- Chronic joint subluxations/dislocations
- Congenital dislocation of the hips
- Talipes equinovarus (clubfoot)
- Gingival recession”
“Probands” is genetic speak for “person presenting now”, vs their parents or children. I actually had Types IV and VI ruled out based on a history of an ascending aortic aneurysm (or AAA) in my dad, facial features, severe scoliosis in my aunt, keratoconus, visible veins, small joint involvement among other things. I was relieved to learn I was negative for both, and we settled on the as yet not fully defined HEDS for my diagnosis.
But that doesn’t mean I don’t still watch for issues, and I did have a baseline echocardiogram done to rule out any aortic root dilation (common in us), or mitral valve issues. (None so far, knock wood). I’ll note I don’t smoke and drink like my parents did, and have led a much healthier lifestyle on average, but that doesn’t preclude issues still. And, as previously noted, there’s nothing very benign about HEDS aside from not dying directly. People with HEDS end up mitigating plenty of serious issues, including severe dysautonomia, weak necks and spines, sciatica, spondylolysthesis and more. (Including anaphylaxis from commonly comorbid MCAD issues, whee).
And I think any and all kinds of EDS can present with aneurysms along the way. Think about it: we’re stretchy folks. So we’ve got notoriously bad “tire walls” all throughout, and every part of us is prone to giving out through herniation or prolapse or tearing, etc. (Dr. Alan Pocinki produced a nice write up in 2010 summarizing the common presentations and signs of HEDS which he still calls Hypermobility Syndrome here). No, it is no cake-walk, though yes, as I’m slowly but steadily proving along with many other hardy survivors, you can recover to some extent through dedicated nutrional work and tailored “zebra-friendly” (core-building) physical therapy that avoids injuring us further, and strengthening the bits we can. I am now walking a bit again thanks to two years of slow, steady hard work in a warm water therapy pool and on my bike, along with disciplined diet and supplementation.
Lastly, I’ll note that having EDS itself, as comprehensive as it is, does NOT preclude having other comorbidities (diseases, conditions or issues), simultaneously. And I mean beyond the incredible list of ones I’ve arleady noted are fairly common in the community (thyroid issues, autism spectrum disorders, depression, CVID, allergies, insomnia, chronic fatigue, fibromyalgia, etc.) I.e, while one woman found some great recovery by addressing her MTHFR defects through nutritional methylation, as my genetics counselor said, this condition may just happen to be true, true, but unrelated, despite Debbie’s hope to the contrary. (MTHFR defects are fairly common in the general population – some cite as high as 40% may have one or more, so it stands to reason many with EDS will have MTHFR defects also – caution, that’s an even DEEPER rabbit hole you can and will get lost in for days if you choose!) You may have any number of other illnesses and genetic disorders also. As someone said, EDS is as individual as fingerprints, and we are all unique individuals in addition espcially when you toss in environmental factors. So while we may share some common “themes” of dysautonomia, fatigue, pain and hypermobility, it will truly express absolutely uniquely within individuals even within the same family. This is known as variable expression. To borrow another phrase from the Autism community: if you’ve seen one Ehlers-Danlos patient, you’ve seen one Ehlers-Danlos patient!
But as you know I feel strongly, every doctor has seen EDS patients whether they know it or not. In fact, everyone has seen an EDSer whether they know it or not, we’re that common IMHO. Hopefully the doctors will start spotting us a bit sooner with this information I’m sharing. (Nurses too!) I hope the above has helped to clarify how best to pursue a diagnosis once you do suspect it.